
Expired activity
Please go to the PowerPak
homepage and select a course.
Module 1. Diabetes Defined: Pathophysiology
INTRODUCTION
Diabetes mellitus has become a disease state of epidemic proportion. This is evidenced by a worldwide
prevalence of at least 8.8%, according to data from the International Diabetes Federation.1 This equates
to a current 425 million people affected worldwide. The global burden of this
disease state is estimated to cause a substantial financial impact in which treatment and possible
prevention strategies will play key roles.
Most simply, diabetes mellitus is characterized by higher than normal levels of glucose in the blood.
Currently, 2 types of diabetes make up the majority of these diagnoses and are known as Type 1 and
Type 2 diabetes.2 Type 1 diabetes is characterized as an autoimmune disease in which the body is unable
to continue to produce insulin because of the destruction of the insulin-producing beta cells in the
pancreas. This type of diabetes typically affects younger individuals who appear to have a thin body
type. Type 2 diabetes is thought to be caused by a genetic predisposition and a complex medley of metabolic risk factors, such as poor diet, obesity, and inactivity. Type 2 diabetes typically occurs in those
older than 30 years of age who are also overweight. Unfortunately, at this time Type 1 diabetes cannot
be prevented. However, there is evidence showing that Type 2 diabetes can at least be delayed, if not
prevented, when at least one risk factor for the disease is eliminated.3,4 Other types of diabetes include
gestational diabetes, genetically induced diabetes, and types that are caused by exocrine; these types of
diabetes are very rare and will only be briefly covered in this module.2 Pharmacists can play a unique
role in the management of diabetes through risk factor modification, medication counseling, and
ensuring that patients diagnosed with any classification of diabetes receive proper therapies.
PREVALENCE
Currently it is estimated that at least 30.3 million people or 9.4% of individuals in the United States (U.S.)
have diabetes.5 In the year 2015 alone, the number of U.S. adults aged 20 years or older newly
diagnosed with diabetes was 1.5 million. Additionally, 23.8% of the more than 30 million people in the
U.S. with diabetes were not yet diagnosed and another 84 million adults (34% of American adults) had
prediabetes. These statistics are staggering and they are projected to continue to grow; estimates
currently suggest that 1 in every 3 Americans born today will develop diabetes. The aging population will
be most impacted, both in the U.S. and in other countries.6 In 2015, diabetes was reported to be the
seventh leading cause of death in the U.S., but this statistic is likely underreported.5 Economically,
$245 billion is spent annually on diabetes in the U.S.1 Our present health care system is likely unprepared to handle the complications
associated with this disease and the massive financial burden it is expected to bring. In addition,
pharmacists and other health care professionals will most certainly be faced with expanding roles in the
management of the symptoms and complications of this disease state.
The following 2 primary types of diabetes comprise the majority of these statistics: Type 1 and Type 2.
The chart below summarizes the similarities and differences among disease states.
| Table 1 - Comparison of Type 1 Diabetes and Type 2 Diabetes7 |
| |
Type 1 Diabetes |
Type 2 Diabetes |
| Phenotype |
Usual onset in children and adolescents |
Usual onset, older than 30 years of age |
| |
Often thin body type or normal BMI (Body Mass Index) |
Often obese |
| |
Prone to ketoacidosis |
Ketoacidosis is seldom spontaneous |
| |
Absolute insulin deficiencies—Insulin required for survival |
Relative insulin deficiency and/or insulin resistance- |
| |
Autoimmune damage to pancreas |
Not autoimmune in nature |
| Genotype |
Identical twin studies: < 50% concordance |
Identical twin studies: usually > 70% concordance |
| |
HLA association: Yes
(human leukocyte antigen) |
HLA association: No
(human leukocyte antigen) |
| |
Increased prevalence in relatives |
Increased prevalence in relatives |
TYPE 1 DIABETES
Epidemiology
Type 1a diabetes mellitus or immune-mediated diabetes, more commonly known as Type 1 diabetes,
comprises only 5% to 10% of all diabetes cases worldwide.2 However, it is one of the most common
chronic childhood disease states. Idiopathic Type 1 diabetes or Type 1b diabetes mellitus occurs when
patients with no evidence of autoimmunity experience complete insulin deficiency; this type will not be
discussed in great detail in this review.8 Type 1 diabetes was previously known as insulin-dependent
diabetes or juvenile-onset diabetes and nearly 500,000 children live with it and almost 79,000 develop
the disease every year worldwide.1 While this impact is very small compared with the number of
individuals with Type 2 diabetes, the incidence of Type 1 diabetes is increasing by approximately 3.2%
per year.9 The reasons for the upward trend are not clear, but it is important to remember that
individuals diagnosed with Type 1 diabetes can live well into their older adult years if the disease is
managed appropriately.
Type 1 diabetes usually presents in children and young adults. For individuals younger than 20 years of
age, Type 1 diabetes accounts for the majority of new diabetes cases diagnosed in the U.S.10 The age of
presentation peaks at the following 2 distinctive points: one peak occurs from 4 to 6 years of age and
the other peaks in early puberty (i.e., 10 to 14 years of age).11 Overall, roughly 45% of children with Type
1 diabetes are diagnosed when they are younger than 10 years of age; however, adults comprise one-quarter of all Type 1 diabetes diagnoses.2
Clinical Presentation
Type 1 diabetes can present in several different ways. Children tend to present with the abrupt onset of
polydipsia, polyuria, weight loss, and hyperglycemia.12 Hyperglycemia sufficient enough to be classified
as diabetes is when glucose exceeds any of the following markers: random plasma glucose of >200
mg/dL with symptoms or fasting plasma glucose of >126 mg/dL or hemoglobin A1c of greater than or equal to 6.5%.7 Children with these symptoms usually appear slightly ill with nonspecific complaints, such as lethargy
and weight loss, although weight loss occurs in only half of children diagnosed.13 Polyuria may present as
bed-wetting or daytime increased urinary frequency and/or incontinence in a previously potty-trained
child. Other symptoms include blurry vision, drowsiness, poor stamina, frequent skin and bladder
infections, and vaginitis. Adults present similarly, but symptoms may not be as abrupt. For adults, longer
time frames from the onset of disease to the diagnosis often occur as the result of a decreased rate of
beta-cell destruction.2 At the time of diagnosis, 80% to 90% of beta cells have already been destroyed in
these patients.
Type 1 diabetes can first present as diabetic ketoacidosis (DKA), which is the most concerning
complication associated with this disease. Approximately, 20% to 40% of patients with Type 1 diabetes
present in DKA and will require hospitalization.2 Children are more likely than adults to present in DKA
and this syndrome is characterized by nausea and vomiting, breathlessness, and abdominal pain.14 Laboratory values will indicate that hyperglycemia, glucosuria, ketonemia, and ketonuria are present. In
addition, patients may also develop a fruity-smelling breath and increased drowsiness and lethargy. DKA
also leads to profound dehydration associated with polyuria and sometimes acute vomiting. DKA in
children is reported to occur approximately 25% to 30% of the time at diagnosis.15 Children younger
than 6 or those from an adverse socioeconomic background are more likely to present in DKA.16 Patients
with DKA will require abrupt treatment, including rehydration and insulin therapy in the hospital
setting.2
After diagnosis, many patients enter a honeymoon phase, in which there is some endogenous insulin
production and only small amounts of exogenous insulin is required to maintain glycemic control.17 This time period is transient and beta-cell destruction continues, leading to a complete reliance on
exogenous insulin for euglycemia.
Pathophysiology
It has long been assumed that Type 1 diabetes results from an interplay between genetics and
environment. More specifically, in a person who is genetically predisposed to the disease,
environmental triggers are thought to cause an autoimmune response that leads to destruction of
pancreatic beta cells (i.e., cells that excrete insulin) and, ultimately, insulin deficiency and metabolic
abnormalities.8 The main autoantigen is unknown, but it may be insulin itself.18 Additionally, there does
appear to be a common cascade of events in the lifeline of the immune-mediated illness. These events
include a long period of time prior to diagnosis in which immune markers are present and beta-cell
destruction is occurring (i.e., reported to be as long as 9 years), high blood glucose levels (during which
80% to 90% of pancreatic beta cells are destroyed), a transient period of remission known as the honeymoon phase (during which the individual appears to produce some insulin) and, finally,
established disease.14
On examination, the involved pancreatic islets contain beta cells with enlarged nuclei, variable numbers
of degranulated beta cells, and a chronic inflammatory infiltrate that is known as insulitis.19 This infiltrate is made of T lymphocyte cells known as CD8 and CD4 and natural killer cells and macrophages.
The involvement of the islets is not uniform. Within the same pancreas, there are areas of complete
destruction of beta cells, islets that are intact but surrounded by inflammation, and islets that are left
with normal function. The variability seen in the damage of the pancreas may help explain why there is a
slow progression to overt hyperglycemia, especially in adults. Healthy beta cells of the pancreas also
produce amylin.20 Amylin is a hormone that slows gastric emptying, improves satiety, and suppresses
glucagon secretion after meal intake. Because the beta cells no longer function, amylin is also not
produced, leading to higher blood sugars after meal intake. Thus, insulin and amylin deficiencies cause
many metabolic abnormalities, including impaired glucose, lipid, and protein metabolism.
| Figure 1 – Pathophysiology of Type 1 Diabetes Timeline19,20 |
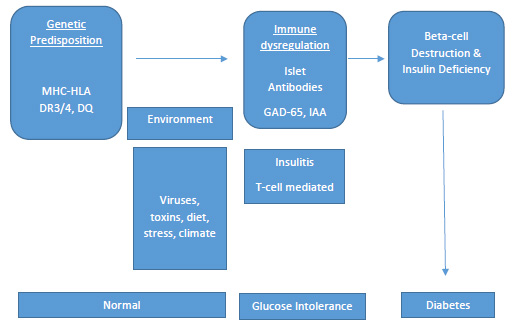 |
| MHC = major histocompatibility complex; HLA = human leukocyte antigen; GAD65 = glutamic acid decarboxylase65; IAA = insulin autoantibodies |
Risk Factors
Family History
Studies show that the lifetime risk of developing Type 1 diabetes is substantially greater for those who
have close relatives with Type 1 diabetes. The risk of Type 1 diabetes in the general population is
reported to be approximately 1 in 400 or 0.25%.21 The risk increases 1% to 8% if either the mother or the
father has Type 1 diabetes and contributes the disease22; the risk is a bit higher if the father contributes
the disease instead of the mother. If both parents are affected with Type 1 diabetes, the risk is
substantially greater and reported to be as high as 30%. Additionally, the sibling risk is similar to the risk
with one parent contributor, at 3% to 6% for non-twins. The incidence increases to as much as 50% for
identical twins.
Race/Ethnicity
Ethnic differences in the incidence of Type 1 diabetes is also apparent in the U.S. The highest prevalence
of Type 1 diabetes was seen in non-Hispanic white youths, according to a 2009 study that sampled large
multiethnic populations.23 Non-Hispanic whites younger than 20 years of age seem to have the highest
incidence of Type 1 diabetes in the U.S., at 23.6 per 100,000 person-years.11 The incidence for African-Americans, Hispanics, Asian-Pacific Islanders, and American Indians followed in respective order, from
rates highest to lowest after non-Hispanic youths. These observations of ethnic-specific risk factors are
likely contributed to gene polymorphisms.
Genetic Markers
Type 1 diabetes is most definitely a polygenic disorder and more than 40 genetic polymorphisms that
confer susceptibility to Type 1a diabetes have been identified.24 Multiple genes are reported to
influence the risk of Type 1 diabetes, including human leukocyte antigen (HLA)-DQalpha, HLA-DQBeta,
HLA-DR, preproinsulin, the PTPN22 gene, CTLA-4, interferon-induced helicase, interleukin (IL)-2 receptor
(CD25), and many others. The genes most responsible for familial aggregation of diabetes are within the
major histocompatibility complex (MHC) located in the HLA region on chromosome-6 and are thought to
be responsible for more than 40% to 50% of Type 1 diabetes genetic susceptibility.25 Certain
combinations of the DQ and DR alleles are associated with a higher risk of Type 1 diabetes. In particular,
more than 90% of patients with Type 1 diabetes carry either HLA-DR3, DQB1*0201 or HLA-DR4,
DQB1*0302.26 Additionally, DR3/4 heterozygotes are at the greatest susceptibility and carry the
DQA1*0501, DQB1*0201, and DQA1*0301, DQB1*0302 gene sequences. These DR3/4 heterozygotes
comprise only 2% of the children born in the U.S.; however, these individuals make up 40% of all U.S.
children that develop diabetes. In addition, some of the HLA alleles are associated with protection from
diabetes including DQA1*0102, DQB1*0602.27 This protective allele occurs in 20% of the general
population, but in less than 1% of children who develop Type 1 diabetes.
Non-HLA genes identified as possibly contributing to the development of Type 1 diabetes include the
insulin gene (INS) region on chromosome 11, polymorphisms of a promoter of the insulin gene, and an
amino acid change of a lymphocyte specific tyrosine phosphatase (PTPN22).28 The association between
these genes and the development of diabetes is not as strong as the HLA association. However, MHC
susceptibility genes are not sufficient to induce Type 1 diabetes alone, suggesting that polygenic
inheritances of non-MHC genes are essential to the development of the disease.27
Autoimmunity
The natural progression of Type 1 diabetes is hypothesized to occur as the result of a combination of
genetic susceptibility factors and environmental triggers causing autoimmunity.2 Autoimmunity in Type
1 diabetes is described as the presence of circulating antibodies to islet and/or beta-cell antigens, often
long before the disease state becomes evident.29 The exact mechanism as to how beta-cell
autoimmunity is initiated after the precipitating environmental factor is not yet proven. Islet cell
autoantibodies have been identified in 85% of patients with newly diagnosed Type 1 diabetes, as well as
in individuals with prediabetes. Several specific autoantigens have been identified that may play
important roles in the initiation of injury to the beta cells, including insulin autoantibodies (IAA), tyrosine
phosphatase islet antigen 2 (IA2A), islet-specific-glucose-6-phosphatase catalytic-subunit-related protein
(IGRP), glutamic acid decarboxylase 65 (GAD65), zinc transporter of islet beta cells (ZnT8) and others.30 The presence of specific autoantibodies can be a useful tool in establishing diagnosis because the risk of
developing Type 1 diabetes is strongly correlated with the number of positive antibodies. However, the
absence of pancreatic autoantibodies does not rule out the possibility of developing Type 1 diabetes and
it is estimated that 5% to 10% of all patients with Type 1 diabetes are not positive for islet
autoantibodies.8 This indicates that other islet autoantigens may exist.
Additionally, patients with Type 1 diabetes are also more likely to be diagnosed with other autoimmune
disorders, including celiac disease, thyroid disorders (e.g., Graves’ disease and Hashimoto’s thyroiditis),
Addison’s disease, multiple sclerosis, and others.2 This suggests that simply having an autoimmune
disorder may put a person at greater risk for Type 1 diabetes as well.
Environmental Associations
Despite multiple research efforts aimed at identifying specific environmental triggers for diabetes, no
clear factor has been linked to islet autoimmunity.17
Geographic location may play a role in predicting the development of Type 1 diabetes. Although the
incidence of childhood diabetes is quite variable across the world, specifically in Europe, the risk appears
to rise in accordance with one’s distance from the equator.31 The highest reported incidences of Type 1
diabetes occur in Finland and Sardinia, with rates almost 400 times higher than those of children in
China and Venezuela, which have the lowest incidence.32,33 However, wide variations in incidence among
locations of similar latitude suggest that other factors, besides location, are major contributors.
Perinatal factors, such as maternal age older than 25 years, preeclampsia, maternal diet, maternal
infections, jaundice, and neonatal respiratory disease, have been shown to be associated with small
increases in risk for developing Type 1 diabetes.34 The observation that Type 1 diabetes autoantibodies
often develop early in infancy suggests that fetal exposures may play a role in future disease
development. More research is needed in this area for definitive causation to be proven.
Both specific childhood virus exposures and a low overall rate of infection in childhood have been
proposed as possible environmental causes of Type 1 diabetes.35 Viruses identified include mumps,
rubella, enteroviruses, rotavirus, parvovirus, and cytomegalovirus. Enteroviruses have an affinity for islet
cells, specifically, and have been isolated from the islets of patients with Type 1 diabetes. However, this
evidence is not conclusive and it is not certain that any of these viruses are involved in causation of the
disease. To further confuse the issue, there is evidence that other viruses may protect against the
development of Type 1 diabetes, which was suggested by a study that showed increased rates of
diabetes in mice that were raised in pathogenic-free environments.36 This theory is similar to the
“hygiene hypothesis” and may have to do with the gut microbiome.
It has also been suggested that proteins in cow’s milk may trigger an autoimmune response.37 This is
either as a result of early exposure to some component of albumin in the milk or from a cell-mediated
response to beta-casein. Longer duration of breast-feeding and supplementation with Vitamin D have
been reported to provide partial protection against beta cell autoimmunity and Type 1 diabetes.This
research is ongoing as other studies have not found an association between increased rates of type 1
diabetes and early exposure to cow’s milk or lesser duration of breast-feeding.38
TYPE 2 DIABETES
Epidemiology
Type 2 diabetes makes up the majority of diabetes diagnoses and represents approximately 90% to 95%
of all diabetes cases worldwide.1 Often referred to as adult-onset diabetes or non-insulin-dependent
diabetes, this disease state is generally diagnosed in adults older than 30 years of age but can be
diagnosed at any age. This epidemic is most certainly linked to increasing rates of overweight and obese
persons in the United States (U.S.) The prevalence of Type 2 diabetes is 3 to 7 times higher for obese
adults compared with adults at an optimal body weight.20 It appears to be most common in both the
older and non-white population of the U.S. As much as 25% of Americans older than 65 years or older are
shown to have diabetes.5 Additional data suggest that the diagnosis of Type 2 diabetes is increasing for the younger population as well. Of the children diagnosed with Type 2 diabetes, 94% are of minority
descent, and being overweight or obese especially increases the risk.
Clinical Presentation
Type 2 diabetes is a disease characterized by high blood sugar, similar to Type 1 diabetes. However,
unlike Type 1 diabetes, the onset of Type 2 diabetes tends to progress gradually. Typical first symptoms
in adults include fatigue, dry skin, poor wound healing, dry mouth, blurred vision, or sometimes there
are no symptoms at all.39 Symptoms tend to be poorly differentiated, but can also present overtly,
similar to those of Type 1 diabetes, especially if the disease has gone undetected for longer periods of
time. The wide range of symptoms at presentation reflects the level of insulin resistance and degree of
beta-cell damage that has occurred. Many people already experience long-term complications at initial
presentation and this has led scientists to estimate that diabetes may typically be present for 4 to 7
years prior to diagnosis. Examples of these complications include neuropathies, retinopathies,
cardiovascular issues, and other microvascular concerns. Because symptoms at presentation vary so
widely and are often not recognized, screening of high-risk persons is critical for identifying those with
prediabetes or undiagnosed diabetes and preventing the disorder if possible. Hyperglycemia that is
pronounced enough to diagnose diabetes is the same criteria used to diagnosis Type 1 diabetes.7 Hyperlipidemia and hypertension are common metabolic comorbidities in these patients and screening
all patients with diabetes for blood pressure and lipid abnormalities at diagnosis is recommended.
Type 2 diabetes in children most often occurs in those diagnosed as overweight or obese and in those
with a family history of the disease. As many as 85% of children diagnosed with Type 2 diabetes are
overweight or obese and as many as 74% to 100% of these children have a first-degree relative with the
disease.40 As mentioned before, many of these children are of non-European descent. In general,
children present with glycosuria but no evidence of ketonuria, mild thirst complaints, possible slight
increase in urination, and little or no weight loss.39 Polycystic ovary syndrome (PCOS) and acanthosis
nigricans (thickening and darkening of skin in the neck, groin, or armpit region) are disorders associated
with insulin resistance and these are commonly seen in children presenting with Type 2 diabetes as well.
The onset of childhood Type 2 diabetes typically occurs around puberty, when insulin sensitivity
declines. Metabolic disturbances are also seen in children newly diagnosed and include hypertension
(10% to 32%), dyslipidemia (18% to 8%), and microalbuminuria (14% to 22%).
Pathophysiology
Type 2 diabetes is a progressive disease caused by multiple complex metabolic factors that result from
defects of specific organ sites. While the mechanism of this process is not completely understood, it is
clear that the following abnormalities are occurring: insulin resistance in muscle and adipose tissue, a
progressive decline in pancreatic insulin secretion, higher glucagon levels, which cause increased hepatic
glucose production, and diminished production of gastrointestinal incretins.2 This process occurs in a
progression of clinically defined phases.
Phase 1 is considered the initial stage of the disease during which insulin resistance first occurs, along
with impaired insulin sensitivity, and is soon followed by compensatory insulin hypersecretion.39 Almost
all patients are asymptomatic at this stage and clinicians are unaware of the disease process. Phase 2 is
when pancreatic beta-cell secretion of insulin is impaired even further, so that an abnormal rise of blood
sugar occurs after meal intake and fasting glucose levels also rise. This phase is typically called prediabetes. Fasting blood glucose typically rises to 100 mg/dL or higher (100 to 125 mg/dL) and this is
referred to as impaired fasting glucose, Upon observation, 2-hour postprandial readings climb to 140
mg/dL or higher (140 to 199 mg/dL) when impaired glucose tolerance occurs and hemoglobin A1c
readings can be found slightly elevated to 5.7% or greater (5.7% to 6.4%).7,20 Lastly, in phase 3, overt
diabetes occurs as the result of a progressive decline in beta-cell function as well as additional decreases
in insulin production. Lack of insulin sensitivity is accompanied by increased hepatic glucose production
as well.39 This phase is sufficient enough to produce fasting glucose levels diagnostic of diabetes (126
mg/dL or greater); however, most will still not experience any of the symptoms of diabetes. It is
important to remember that progression to diabetes among those with risk factors for diabetes is not
inevitable. Several studies, including the Diabetes Prevention Program Research Group, have shown that
people with prediabetes who exercise regularly and lose weight can at least delay, if not prevent Type 2
diabetes.4
| Figure 2 – Natural Progression of Type 2 Diabetes.41 |
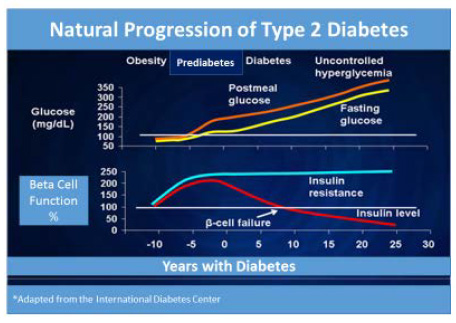 |
Insulin Resistance
Type 2 diabetes is a complex syndrome characterized by the following 2 defective insulin-mediated
processes: impaired glucose metabolism into insulin-sensitive tissues and the decreased effectiveness of
insulin to suppress glucose production by the liver.42 Multiple cellular defects in insulin action have been
discovered, including impairment of glucose transport, glucose phosphorylation, glycogenesis, and
glucose oxidation. Central obesity appears to be one of the most important causes of insulin resistance
development. Insulin resistance also contributes to lipid abnormalities, as evidenced by elevations in
triglyceride-rich lipoproteins and decreases in high density lipoprotein (HDL) cholesterol levels for those
individuals with hyperinsulinemia. When a patient possesses multiple risk factors for heart disease,
including central obesity, elevated lipids, increased blood glucose, and/or hypertension; this myriad of
risk factors is called metabolic syndrome.2,43 These patients are at especially high risk for diabetes and end organ complications. Epidemiological studies suggest that high insulin levels may be associated with
coronary artery disease, as well, and that insulin resistance correlated with carotid intimal medial wall
thickness.43 A more precise measure of insulin action is critical for defining the relation between insulin
resistance and coronary artery disease.
Beta-Cell Dysfunction
Normally, pancreatic beta cells respond to changes in blood glucose and food intake in a complex
fashion that maintains normal blood glucose levels throughout the day.20 Type 2 diabetes impairs beta
cell function in several ways. When insulin resistance and increased hepatic glucose output are present,
initially pancreatic insulin secretion is increased to compensate for these impairments temporarily.
However, persistent fasting hyperglycemia does not allow this compensatory mechanism to continue
forever and eventually insulin secretion declines. About 70% of beta-cell function is already lost by the
time the 2-hour postprandial glucose level is 120-140 mg/dL.44 Some studies show that a decrease in the
beta-cell mass and an increase in beta-cell apoptosis may be the causation of impaired insulin secretion
and an overall decrease in beta-cell function.45 Individuals with prediabetes and Type 2 diabetes have
been shown to have decreased beta-cell mass compared with their counterparts without diabetes,
regardless of weight status.
Hepatic Glucose Production
A secondary cause of increasing blood glucose levels is excessive hepatic glucose production.2 In fasting
conditions, glucose is produced by the liver; however, the liver of a person with diabetes will produce
substantially more glucose and burden the circulation with extra glucose. This is caused by the excess
glucagon levels of people with Type 2 diabetes. Reduced insulin/incretin hormones cannot allow for the
normal suppression of glucagon levels in hyperglycemic conditions. In addition, insulin normally causes
glucose uptake into the muscle and adipose tissue and a decrease in hepatic glucose production. Insulin
is needed to signal the liver to stop glucose production; so, as insulin levels decline, the liver produces
more glucose than the body can use and glucose levels in the plasma continue to rise.
Incretin Effect
Incretin hormones in the gastrointestinal tract have been implicated as a factor in the development of
Type 2 diabetes. Incretins are naturally occurring hormones that the gut releases throughout the day to
facilitate the response of the pancreas and liver to variations in blood glucose.46 The main incretins
responsible for this are thought to be glucose-dependent insulinotropic peptide (GIP) and glucagon-like
peptide-1 (GLP-1). The activity level of these incretins is increased substantially when food is ingested.
When blood glucose rises, both GIP and GLP-1 stimulate beta cells to secrete insulin and the alpha cells
of the pancreas to suppress glucagon release from the liver. Patients with prediabetes and Type 2
diabetes are deficient in GLP-1. This deficiency contributes to excess hepatic glucose production, failure
to suppress production of hepatic glucagon after meal intake, and increased appetite. These same
people also show increases in dipeptidyl peptidase-4 activity (DPP-4) in the fasting state. DPP-4 enzymes
break down GLP-1 hormones in the body and this may explain why GLP-1 secretion is impaired after
meal intake in those with Type 2 diabetes.20
| Figure 3. Contributing Factors in the Pathophysiology of Type 2 Diabetes2,20,42 |
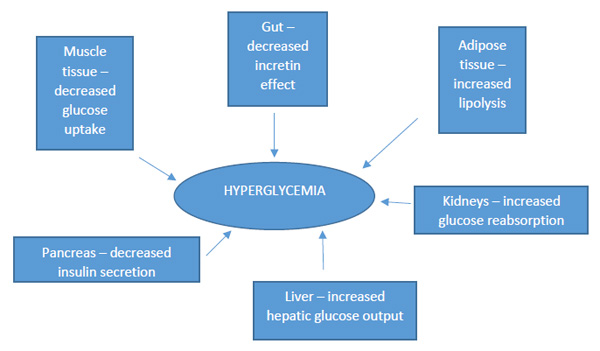 |
RISK FACTORS
Family History/Ethnicity
The development of Type 2 diabetes is clearly linked to family history. In fact, at least 39% of patients
with Type 2 diabetes have at least one family member with the disease.47 Additionally, Type 2 diabetes
prevalence varies tremendously among groups with different ethnic origins, with non-white Americans
of African-American, Native American, and Hispanic origin having 2 to 6 times the rate of Type 2
diabetes as compared with whites living in the same environment.48 First-degree relatives of patients
with Type 2 diabetes are at least 3 times more likely to develop the disease than those without a family
history of the disorder.39 Very high concordance rates, some as high as 90% (i.e., Type 2 diabetes
presenting in monozygotic twins), suggest that the heritability of Type 2 diabetes exists.49 This clustering
of Type 2 diabetes within family lines is complicated by other factors, such as shared environmental risk
factors, like obesity and sedentary lifestyle, making family history likely only partially responsible.
Furthermore, Type 2 diabetes is much more common than Type 1 diabetes and the appearance of
subsequent family members with the disease may just reflect that commonality.
Genetics
The genetics of Type 2 diabetes is quite complex and not completely understood. Most cases of Type 2
diabetes involve many genes contributing small parts to the overall condition. More than 36 genes have
been identified as contributing to risks for Type 2 diabetes, but only 10% of the heritability of Type 2
diabetes can be explained.20 For most patients, it is impossible to identify a genetic abnormality and
environmental factors are more predominate. The genes that have been associated with Type 2 diabetes include transcription factor 7-like 2 (TCF7L2), peroxisome proliferator-activated receptor
gamma (PPARG), potassium inwardly rectifying channel - subfamily J - member 11 (KCNJ11), the
sulfonylurea urea receptor (ABCC8), Calpain 10, glucose transporter 2 (GLUT 2), the glucagon receptor
(GCGR), and many others.50 Defects at TCF7L2 impact beta-cell development and function and studies
show that carriers of one risk allele have an approximate risk of Type 2 diabetes that is 40% higher than
carriers with a protective allele. Additionally, one genetic variant of the PPARG gene causes decreased
insulin sensitivity and increases the risk for Type 2 diabetes by severalfold. This gene is thought to be
more common in Caucasians. Calpain 10 and ABCC8 variations contribute to decreased insulin secretion
and, thus, increase risk for diabetes. Interestingly, most of the genes identified impact insulin secretion
and regulation mediated from the pancreas. In contrast, genes that have been linked to insulin
resistance, obesity, and other aspects of glucose metabolism are much less common.
Environmental Factors
Obesity, aging, a longer life span, smoking, insufficient energy consumption, and physical inactivity are
all environmental factor examples, affecting the progression of the Type 2 diabetes continuum.51 Central
or visceral obesity seems to be specifically problematic and a major contributor to insulin resistance.
Decreased daily activity related to modern technology and changes in the American diet, including
increased fat intake, increased simple sugar intake, and a decrease in dietary fiber intake, have all
contributed to the obesity epidemic as well. Even mild obesity can contribute to glucose intolerance.
Those of Japanese origins are especially prone to central obesity and, thus, a lower body mass index
(BMI) target has been recently suggested for them (23 BMI).7 Smoking has been shown to be
especially detrimental to health and increases the risk for diabetes by 30% to 40% , along with the long-term complications risk.52
Medications that may impair glucose tolerance by decreasing beta-cell insulin secretion, increasing
glucose production in the liver, or causing insulin resistance are vast. Glucocorticoids, oral
contraceptives, beta-blockers, thiazide diuretics, statins, protease inhibitors, atypical antipsychotics,
cyclosporine, tacrolimus, and others comprise this list.53 Pharmacists should be especially vigilant to
review all medications for patients with diabetes, or those at risk for diabetes, for possible
hyperglycemia adverse reactions.
Intrauterine developmental causes have also been implicated in the development of Type 2 diabetes
recently. The theory of the thrifty genotype suggests that insulin resistance is both beneficial and
detrimental by improving survival during states of caloric deprivation, but leading to negative
consequences in states of caloric excess.54 This theory has been hypothesized to occur during early life in
the womb if intrauterine growth restriction and low birth weight are caused by malnutrition. Thus, those
infants born with low birth weights (less than 3.5 kg) are at greater risk for developing diabetes in
adulthood. Conversely, high birth weight is also a risk factor for diabetes (greater than 4 kg).55 It has
been shown that prenatal exposure to hyperglycemia can cause higher birth weights and may increase
the risk for Type 2 diabetes, regardless of genetic predisposition. Other studies indicate that premature
children may also be at greater risk for diabetes.55 More research is needed to confirm these findings.
However, it is apparent that prenatal influences may have great determinants for the risk of developing
diabetes in the future.
OTHER TYPES OF DIABETES
Latent autoimmune diabetes in adults (LADA), also known as Type 1.5 diabetes, is a slow, progressive
form of Type 1 diabetes that is often misdiagnosed as Type 2 diabetes initially.20 Even though these
patients experience the same autoimmune process as those with the classic presentation of Type 1
diabetes, they may not have to inject insulin treatments for many years because of the slow onset of
disease. Patients with LADA are typically not obese and are usually older than 35 years of age. They may
be able to control blood sugars initially with diet and then progress to oral agent use; however,
inevitably, they will require insulin at some point. The time frame from the initial diagnosis of LADA to
the use of insulin is variable, depending on the degree of beta-cell dysfunction present. Most patients
with LADA will require insulin soon after diagnosis, but some may be able to function without insulin use
for longer periods of time. Because of this, LADA is often misdiagnosed as Type 2 diabetes initially.
Idiopathic diabetes is a form of Type 1b diabetes with no known etiology.7 Patients with this disorder are
prone to ketoacidosis and exhibit varying degrees of insulin deficiency, but have no evidence of
autoimmunity. This form of the disease appears to be strongly tied to family inheritance and most of
those affected by it are of Asian or African ancestry.
Maturity-onset diabetes of the young (MODY) is a rare form of diabetes that results from a defect in a
single gene.7,20 Abnormalities at 6 genetic loci on different chromosomes have been identified as
contributors. Monogenic in nature and very rare, this type of diabetes only accounts for 1% to 5% of all
diabetes diagnoses in young people. MODY does tend to run in families and typically occurs in
adolescence or early adulthood. People with MODY are usually not overweight and do not present with
other risk factors for diabetes. MODY can be treated with oral antihyperglycemic agents and often does
not require insulin.
Gestational diabetes is termed as any degree of glucose intolerance with onset during pregnancy.7 The
incidence of gestational diabetes varies among populations and ethnicities; but, it is estimated that of
the 6% to 7% of pregnancies affected by diabetes, 90% of them are gestational diabetes. The rate of
gestational diabetes is increasing and this is most likely a result of the increasing number of overweight
and obese women in their childbearing years.57 Physical activity and a well-balanced diet are key
counseling points when discussing how to reduce newborn complications for patients with gestational
diabetes. In addition, some expectant mothers may have glycemic levels sufficient enough to warrant
medication therapies. Insulin, sulfonylurea, and metformin are the only medicinal therapies used by
women with gestational diabetes. The risks posed by the medicinal therapy chosen should not outweigh
the benefits of lowering glucose levels. Women who are diagnosed with gestational diabetes are also at
greater risk for Type 2 diabetes in their lifetime and should be monitored regularly after delivery of the
child.
Neonatal diabetes occurs within the first 6 months of life and is extremely rare.7 It has not been shown
to be an autoimmune process like Type 1a diabetes. This type of diabetes can be transient or permanent
depending on the genetic defect. If it is permanent, treatment options depend on the genetic defect and
include sulfonylurea options and/or insulin therapies.
Cystic fibrosis-related diabetes is the most common comorbidity for people with cystic fibrosis.2,7 Partial destruction of the islet mass causes decreased insulin production in this population and nearly 40% to 50% of adults with cystic fibrosis are affected by this condition. These patients are generally treated with
insulin.
Various other secondary forms of diabetes exist; however, they are quite rare and make up less than 2%
of all diabetes statistics.7
SUMMARY
Diabetes is an increasing concern within the U.S., as well as the world. Its remarkable growth rate has
reached an epidemic proportion in recent years. While the majority of diabetes cases diagnosed in the
U.S. are of Type 2 diabetes, cases of Type 1 diabetes are also increasing.1,5 It is difficult to pinpoint the
exact causation for diabetes, but it is clear that environmental exposures, lifestyle factors, and genetics
all play an integral part. Regardless of the etiology, all types of diabetes are associated with long-term
complications. Disappointing data show that hemoglobin A1c values tend to increase by 1% every 2
years, even with the most aggressive interventions.58 Patients with diabetes require repeated
intervention and compliance to therapies for success. Pharmacists in clinical roles can be advocates for
evidence-based practices to decrease the clinical inertia related to diabetes treatment as well as an
important point of contact to patients for medication counseling and adherence assistance. Additionally,
future attempts to prevent and/or reverse diabetes are most likely to succeed if they include the most
recent advances in the understanding of the complex pathogenesis of the disease. Pharmacists play an
important role communicating medical information to patients about diabetes and need to stay updated
on new prevention techniques, treatment devices, and therapies.
| Potential Counseling Tips for Pharmacists |
| Category |
Potential Counseling Tips |
| Prevention |
- Research has shown that the development of type 2 diabetes can be delayed or prevented by eating healthier foods, exercising, or losing weight if necessary.4
- Preventing diabetes in you and your loved ones will not only lead to better health, but will also help you avoid hassle (i.e., less visits to the physician and less time in the hospital) and costs (i.e., costs of medical care and lost work).
- You can potentially prevent diabetes complications by staying in good control.3 This is a great point to bring out to your patients.
|
| Screening |
- Because symptoms at presentation vary so widely and are often not recognized, screening of high-risk persons is critical. The pharmacist can then offer to screen the patient or refer him/her to the next health fair, during which screenings will be performed at low or no cost.
- Patients diagnosed with diabetes type 2 should also receive screenings for high cholesterol and blood pressure.
- Know how to identify the symptoms of diabetes in your patients. Some common symptoms to watch out for are increased thirst, increased urination, and unexplained weight loss.
|
REFERENCES
- Cho NH, Shaw JE, Karuranga S, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018 Apr;138:271-281.
- Triplitt CL, Reasner CA. "Diabetes Mellitus." In: Dipiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 8th ed. New York, NY: McGraw-Hill Medical; 2011.
- The Diabetes Control and Complications Trial (DCCT) Research Group. The effect of intensive treatment of diabetes on the development and progression of long term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977-986.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; for the Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
- Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2017. Atlanta, GA: Centers for
Disease Control and Prevention, U.S. Dept of Health and Human Services; 2017.
- Whiting DR, Guariguata L, Weil C, Shaw J. IDF Diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94(3):311-321.
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2015;38(suppl 1):S8-S16.
- Chiang JL, Kirkman MS, Laffel LM, et al; and the Type 1 Diabetes Sourcebook Authors. Type 1 diabetes through the life span: a position statement of the American Diabetes Association. Diabetes Care. 2014;37(7):2034-2054.
- Patterson CC, Dahlquist GG, Gyürüs E, et al; for the EURODIAB Study Group. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet. 2009;373(9680):2027-2033.
- Duncan GE. Prevalence of diabetes and impaired fasting glucose levels among US adolescents: National Health and Nutrition Examination Survey, 1999-2002. Arch Pediatr Adolesc Med. 2006;160(5):523-528.
- Dabelea D, Bell RA, D'Agostino RB Jr, et al; and the Writing Group for the SEARCH for Diabetes in Youth Study Group. Incidence of diabetes in youth in the United States. JAMA. 2007;297:2716-2724.
- Banion C, Valentine V. "Type 1 diabetes throughout the life span." In: Mensing CE, Cornell S, Halstenson C. The Art and Science of Diabetes Self-Management Education Desk Reference. 3rd ed. Chicago, IL: American Association of Diabetes Educators (AADE); 2014.
- Silverstein J, Klingensmith G, Copeland K, et al; American Diabetes Association. Care of children and adolescents with type 1 diabetes. Diabetes Care. 2005;28(1):186-212.
- Beisswenger PJ. "Type 1 diabetes." In: Leahy JL, Clark NG, Cefalu WT. Medical Management of Diabetes Mellitus. New York, NY: Marcel Dekker; 2000.
- Agus MS, Wolfsdorf JI. Diabetic ketoacidosis in children. Pediatr Clin North Am. 2005;52(4):1147-1163.
- Rewers A, Chase HP, Mackenzie T, et al. Predictors of acute complications in children with type 1 diabetes. JAMA. 2002;287(19):2511-2516.
- Simmons K, Michels AW. Lessons from type 1 diabetes for understanding natural history and prevention of autoimmune disease. Rheum Dis Clin North Am. 2014;40(4):797-811.
- Thrower SL, Bingley PJ. Prevention of type 1 diabetes. Bri Med Bull. 2011;99:73-88.
- Ozougwa JC, Obimba KC, Belonwu CD, Unakalamba CB. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J Physiology and Pathophysiology. 2013;4(4):46-57.
- Bardsley JK, Magee MF. "Pathophysiology of the metabolic disorder." In: Mensing C, Cornell S, Halstenson C. The Art and Science of Diabetes Self-Management Education Desk Reference. 3rd ed. Chicago, IL: American Association of Diabetes Educators (AADE); 2014.
- Haller MJ, Atkinson MA, Schatz D. Type 1 diabetes mellitus: etiology, presentation, and management. Pediatr Clin North Am. 2005;52(6):1553-1578.
- Tillil H, Köbberling . Age-corrected empirical genetic risk estimates for first-degree relatives of IDDM patients. Diabetes. 1987;36(1):93-99.
- Dabelea D, Mayer-Davis EJ, Saydah S, et al; and the SEARCH for Diabetes in Youth Study. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311(17):1778-1786.
- Barrett JC, Clayton DG, Concannon P, et al; and the Type 1 Diabetes Genetics Consortium. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Rev Genet. 2009;41(6):703-707.
- Redondo MJ, Eisenbarth GS. Genetic control of autoimmunity in Type 1 diabetes and associated disorders. Diabetologia. 2002;45(5):605-622.
- Tisch R, McDevitt H. Insulin dependent diabetes mellitus. Cell. 1996;85(3):291-297.
- Erlich H, Valdes AM, Noble J, et al; and the Diabetes Genetics Consortium. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57(4):1084-1092.
- Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet. 2011:12(11):781-792.
- Atkinson MA, Maclaren NK. Mechanisms of Disease: the pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med. 1994;331(21):1428-1436.
- Watkins RA, Evans-Molina C, Blum JS, DiMeglio LA. Established and emerging biomarkers for the prediction of type 1 diabetes: a systematic review. Transl Res. 2014;164(2):110-121.
- Rosenbauer J, Herzig P, von Kries R, et al. Temporal, seasonal, and geographic incidence patterns of type 1 diabetes mellitus in children under 5 years of age in Germany. Diabetologia. 1999;42(9):1055-1059.
- Yang Z, Wang K, Li T, et al. Childhood diabetes in China. Enormous variation by place and ethnic group. Diabetes Care. 1998;21(4):525-529.
- Harjutsalo V, Sund R, Knip M, Groop PH. Incidence of type 1 diabetes in Finland. JAMA. 2013;310(4):427-428.
- Stene LC, Magnus P, Lie RT, et al; Norwegian Childhood Diabetes Study Group. Birth weight and childhood onset type 1 diabetes: population based cohort study. BMJ. 2001;322(7291):889-892.
- Bortell R, Pino SC, Greiner DL, et al. Closing the circle between the bedside and the bench: Toll-like receptors in models of virally induced diabetes. Ann N Y Acad Sci. 2008;1150:112-122.
- Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911-920.
- Knip M, Akerblom HK. Early nutrition and later diabetes risk. Adv Exp Med Biol. 2005;569:142-150.
- Sipetic S, Vlajinac H, Kocev N, et al. Early infant diet and risk of type 1 diabetes mellitus in Belgrade children. Nutrition. 2005;21(4):474-479.
- Vivian EM. "Type 2 diabetes throughout the life span." In: Mensing C, Cornell S, Halstenson C. The Art and Science of Diabetes Self-Management Education Desk Reference. 3rd ed. Chicago, IL: American Association of Diabetes Educators (AADE); 2014
- American Diabetes Association. Type 2 diabetes in children and adolescents (consensus statement). Diabetes Care. 2000;23(3)381-389.
- Bergenstal R. "Natural History of Type 2 Diabetes" diagram. In: Translating Diabetes Medications into Protocol. The Many Faces of Community Health conference. International Diabetes Center (IDC) Web site. http://www.manyfacesconference.org/conference2008/Bergenstal%20Many%20Faces.pdf. Accessed January 12, 2016.
- Leahy JL. "Type 2 diabetes mellitus." In: Leahy JL, Clark NG, Cefalu WT. Medical Management of Diabetes Mellitus. New York, NY: Marcel Dekker; 2000.
- Cefalu WT. "Insulin resistance." In: Leahy JL, Clark NG, Cefalu WT. Medical Management of Diabetes Mellitus. New York, NY: Marcel Dekker; 2000.
- Nichols GA, Hilliar TA, Brown JB. Progression from newly acquired impaired fasting glucose to type 2 diabetes. Diabetes Care. 2007;30(2):228-233.
- Butler AE, Janson J, Bonner-Weir S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102-110.
- Deacon CF. What do we know about the secretion and degradation of incretin hormones? Regul Pept. 2005;128(2):117-124.
- Klein BE, Klein R, Moss SE, Cruickshanks KJ. Parental history of diabetes in a population-based study. Diabetes Care. 1996;19(8):827-830.
- Carter JS, Pugh JA, Monterrosa A. Non-insulin-dependent diabetes mellitus in minorities in the United States. Ann Intern Med. 1196;125(3):221-232.
- Newman B, Selby JV, King MC, et al. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia. 1987;30(10):763-768.
- Herder C, Roden M. Genetics of type 2 diabetes: pathophysiologic and clinical relevance. Eur J Clin Invest. 2011;41(6):679-692.
- Sullivan PW, Morrato EH, Ghushchyan V, et al. Obesity, inactivity, and the prevalence of diabetes and diabetes-related cardiovascular comorbidities in the U.S., 2000-2002. Diabetes Care. 2005;28(7):1599-1603.
- U.S. Department of Health and Human Services. 2014 Surgeon General's Report: The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention (CDC), National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2014. CDC Web site. http://www.cdc.gov/tobacco/data_statistics/sgr/50th-anniversary/index.htm. Accessed November 20, 2015.
- Luna B, Feinglos MN. Drug induced hyperglycemia. JAMA. 2001;286(16):1945-1948.
- Phillips DI, Barker DJ, Hales CN, et al. Thinness at birth and insulin resistance in adult life. Diabetologia. 1994;37(2):150-154.
- Harder T, Rodekamp E, Schellong K, et al. Birth weight and subsequent risk of type 2 diabetes: a meta-analysis. Am J Epidemiol. 2007;165(8):849-857.
- Hofman PL, Regan F, Jackson WE, et al. Premature birth and later insulin resistance. N Engl J Med. 2004;351(21):2179-2186.
- American College of Obstetricians and Gynecologists. Committee opinion no. 504: Screening and diagnosis of gestational diabetes mellitus: College publications retraction. Obstet Gynecol. 2013;122(2 pt 1):405.
- Fonseca VA. Defining and Characterizing the progression of type 2 diabetes. Diabetes Care. 2009;32(suppl 2):S151-S156.
Back to Top