
Expired activity
Please go to the PowerPak
homepage and select a course.
Improving the Impact of the Specialty Pharmacist in the Care of Patients with Acquired Hemophilia A
INTRODUCTION
Hemophilia is a bleeding disorder that impairs the body’s ability to clot blood; this results in longer bleeding after injury, easy bruising, and a higher likelihood of internal bleeding. Two main types of hemophilia exist: hemophilia A and hemophilia B. Hemophilia A is a deficiency in clotting factor VIII, and hemophilia B is associated with a deficiency in clotting factor IX.1 These bleeding disorders can either be inherited or acquired. Inherited hemophilia A and hemophilia B are a result of mutations on the factor VIII and factor IX genes, respectively, which are located on the long arm of the X chromosome. This hereditary disorder occurs almost exclusively in males, with a prevalence of 1 in 5000 men.
Acquired hemophilia, on the other hand, is rare, with an incidence of approximately 1.5 cases per 1 million persons each year: it occurs in men and women with similar frequency.2 Some evidence suggests that the incidence may be underreported because individuals with undiagnosed acquired hemophilia may only be diagnosed if they undergo a surgery or experience trauma. Unlike congenital hemophilia, acquired hemophilia is caused by the spontaneous production of circulating autoantibodies, most commonly autoantibodies against factor VIII. Thus, acquired hemophilia is typically termed acquired hemophilia A (AHA).
The incidence of AHA increases with age: it is more common in adults than in children. The incidence is high in people between the ages of 20 and 30 years, mostly due to autoantibodies that are produced in women during the postpartum period. The incidence is highest in elderly patients: in fact, the median age of diagnosis is 64 to 78 years old, and more than 80% of patients with AHA are aged 65 years or older.2,3 Pediatric cases of AHA are rare, with only 42 cases reported worldwide since tracking began.3 The mortality rate of AHA is estimated to be between 7.9% and 22% of patients, with most hemorrhagic deaths occurring within the first few weeks after diagnosis.
In AHA, the development of factor VIII autoantibodies results in factor VIII depletion, ultimately leading to insufficient generation of thrombin by factor IXa and the factor VIIIa complex via the intrinsic coagulation cascade pathway (Figure 1).4,5 Autoantibody development against factor IX can occur, but this is uncommon. Autoantibodies against other clotting factors are extremely rare. In AHA, autoantibodies are directed specifically at the epitopes of factor VIII, either neutralizing them or causing accelerated clearance from plasma.6 Most commonly, the autoantibodies bind to amino acids in the A2 and A3 domains of the heavy chain of factor VIII and the C2 domain on the light chain of factor VIII.7 Antibodies against A2 and A3 then impede the binding of factor VIII to activated factor X and factor IX pathways; antibodies to C2 inhibit the binding of factor VIII to phospholipids and interfere with factor VIII binding to von Willebrand factor protein.8 Typically, the development of autoantibodies causes rapid declines in factor VIII levels, followed by a slower inactivation, which may result in some residual factor VIII present upon laboratory evaluation. However, functionally, the residual factor VIII is not clinically useful.
Figure 1. Coagulation Cascade5
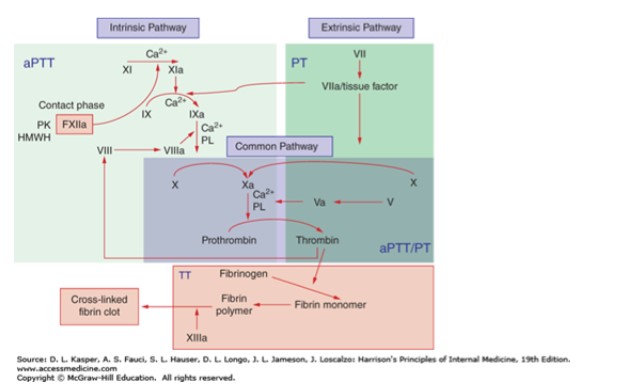
Abbreviations: aPPT, activated partial thromboplastin time; HMWH, high molecular weight heparin; PK, prekallikrein; PL, phospholipid; PT, prothrombin time; TT, thrombin time. |
The pathogenesis of the autoantibody development is not well understood.9 It appears that the spontaneous production of neutralizing antibodies, specifically neutralizing immunoglobulin G antibodies, targets the endogenous production of factor VIII. Both genetic and environmental factors have been implicated as possible triggers.9,10
RISK FACTORS FOR AHA
Most cases (roughly 52%) of AHA are idiopathic, but it can also be associated with other concomitant diseases.3 In females of childbearing age, AHA can occur following pregnancy, typically within 1 to 4 months postpartum.2,3 Thus, women in the early postpartum period are at increased risk for developing AHA (Table 1).2,6 Other conditions that place individuals at risk for the development of AHA include cancers, both hematologic cancers such as leukemia and lymphoma and solid tumors such as lung, prostate, breast, and pancreatic cancers. Cancer is present at the time of diagnosis of AHA in 6% to 18% of patients. Autoimmune disorders, such as rheumatoid arthritis, systemic lupus erythematosus, and multiple sclerosis, can be associated with 9% to 17% of cases of AHA. Less commonly, infections, such as hepatitis B and C, and dermatologic conditions, such as psoriasis and pemphigus, can also be present during AHA. Several medications are also associated with AHA (Table 1).6 In patients with drug–induced AHA, autoantibody titers are typically high (mean 67.7 Bethesda units [BU]/mL). Finally, age is also a risk factor, and patients older than 65 years are at highest risk of developing AHA.
| Table 1. Risk Factors for Acquired Hemophilia A2,6 |
Autoimmune diseases
Systemic lupus erythematosus, rheumatoid arthritis, temporal arteritis, ulcerative colitis, Sjögren's syndrome, Goodpasture syndrome, multiple sclerosis, myasthenia gravis, autoimmune thyroid disorders, autoimmune hemolytic anemia |
Dermatologic diseases
Psoriasis, pemphigus |
Cancer
Hematologic: chronic lymphocytic leukemia, multiple myeloma, Waldenström macroglobulinemia, non-Hodgkin lymphoma, myelodysplasia, myelofibrosis, erythroleukemia
Solid: lung, prostate, pancreas, colon, stomach, melanoma, breast, kidney, cervix, head, neck |
Infections
Acute hepatitis B, acute hepatitis C |
Increasing age
Older than 65 years |
Medication use
Beta-lactam antimicrobials, chloramphenicol, clopidogrel, fludarabine, interferon-alpha, methyldopa, nonsteroidal anti-inflammatory drugs, phenytoin, sulfa drugs, thioxanthenes (depot preparations) |
Other diseases
Chronic obstructive pulmonary disease, asthma, monoclonal gammopathy of uncertain significance |
Women in postpartum period
1-4 months after childbirth |
CLINICAL PRESENTATION AND DIAGNOSIS OF AHA
Approximately 95% of patients with AHA will present with a bleeding episode. Spontaneous bleeding will occur in 77% of patients, and 70% to 90% of patients will have a serious bleed, resulting in a decrease in hemoglobin by 2 g/dL or to a level less than 8 g/dL and the need for hemostatic transfusion support.2,3 The most common sites of bleeding are in the mucous membranes and skin (80%), muscles (> 40%), and gastrointestinal areas (> 20%).2 Spontaneous bruising, epistaxis, and gastrointestinal, retroperitoneal, urologic, and muscle hematomas are also common. Another common presentation is the development of iatrogenic bleeding after attempts to insert intravenous lines. Less frequent manifestations are prolonged postpartum bleeding, excessive bleeding following trauma and surgery, and, rarely, cerebral hemorrhage. Unlike in congenital hemophilia, intra–articular bleeding is uncommon in AHA.
The bleeding symptoms are often serious and life threatening, and, thus, AHA is considered a severe bleeding disorder associated with significant morbidity.3 Because the severity of bleeding often does not correlate with factor VIII levels or inhibitor titers, clinicians must treat the bleeding, regardless of the laboratory results.11 Mortality is estimated to be 20% or greater among patients 65 years and older and among those with cancer. However, most patients will die of their underlying disease (46%), rather than AHA or the bleed itself. Significant morbidity often occurs and results in diagnostic delays, inadequate treatment, bleeding complications, extended hospitalizations, and healing delays. Indirectly, these may result in death due to complications that arise during these delays and hospitalizations.
Thus, AHA should be considered in patients without a personal or family history of a bleeding disorder and with a sudden onset of bleeding that is often severe, occurring spontaneously, and occurring after minor trauma or following invasive procedures or surgery. In particular, AHA should be suspected in those within the setting described above (i.e., hospitalized and experiencing bleeding complications and healing delays), in women who are in the peripartum or postpartum period, and in elderly patients.2,3
In patients with these symptoms, the platelet count, bleeding time, and prothrombin time (PT) are normal, but the activated partial thromboplastin time (aPTT) is prolonged.2 To determine if a simple factor deficiency or inhibitor development exists, a mixing study (also called a correction test) is conducted. A mixing test is done by mixing the patient plasma in a 1:1 ratio with plasma that contains 100% of the normal factor level, resulting in a level of at least 50% in the mixture. With a factor deficiency, the PT or aPTT will be normal (i.e., the mixing study shows correction). However, with inhibitor development such as occurs in AHA, a prolonged PT or aPTT is not reversed using a mixing study (i.e., the mixing study fails to correct). A prolonged aPTT can also be caused by the presence of a lupus anticoagulant or the use of medications such as heparin, low molecular weight heparins, direct factor Xa inhibitors (e.g., rivaroxaban, apixaban, edoxapan), direct thrombin inhibitors (e.g., argatroban,
dabigatran), fondaparinux, and, rarely, high doses of warfarin. Thus, if a patient requires one of these drugs—even a heparin flush—the medication must be stopped for a long enough time to ensure that it is no longer circulating in the plasma, and the aPTT must be redrawn in order to obtain an accurate result. Similarly, a lupus anticoagulant test needs to be completed and the result needs to be negative in order to move forward with a diagnosis of AHA.
If the lupus anticoagulant is negative and aPTT continues to be prolonged in the absence of medications that cause prolonged aPTT, then AHA can be confirmed by measuring the factor VIII coagulant activity. Typically, the Bethesda assay is used to measure factor VIII coagulant activity. One BU is the quantity of inhibitor that inactivates 50% of factor VIII in normal plasma after incubation at 37 °F for 2 hours. AHA is categorized by low activity (< 5 BU/mL) or high activity (> 5 BU/mL).2 The Nijmegen modification to the assay minimizes shifts in pH and allows for better detection at low activity levels.8 More recently, a commercially available ELISA assay became available that is able to detect anti–factor VIII immunoglobulin G autoantibodies with similar specificity and sensitivity to historic tests: it is also less expensive and easier to interpret.2
CLINICAL MANAGEMENT OF AHA
The goals of AHA management are to eliminate the underlying cause of AHA, if possible, control acute bleeding episodes, and ensure long–term eradication of the autoantibodies.2,8 Two main therapeutic approaches are used: 1) hemostatic agents, which are agents that control the hemostasis and prevent and/or resolve bleeding episodes; and 2) eradication therapy, which is therapy aimed at removing the autoantibodies from the body. If possible, a hematology specialist familiar with treating AHA should care for these patients.3 Hemostatic agents often do not have predictable efficacy and underlying comorbidities can place patients at risk for thrombotic events. Furthermore, the immunosuppressive therapy that might be used to eradicate autoantibodies typically causes an unstable hemostatic environment. Thus, both a specialist and a facility that have expertise, access to these agents, and capabilities to provide timely monitoring are of utmost importance.
Hemostatic therapy
Although avoidance of surgery and interventional procedures is desired in AHA, at times, patients may need emergent surgery or an alternative to surgical intervention may not be available. In these cases, patients will receive hemostatic therapy before the procedure.3 More commonly, however, hemostatic agents are administered to patients with acute bleeding episodes or to those at high risk for a bleed (e.g., recent surgery, childbirth, gastrointestinal ulcers). In those with a major bleeding episode and/or a decrease in hemoglobin, hemostatic agents are administered immediately; those with mild–to–moderate bleeding but no significant decrease in hemoglobin may not require immediate administration. The decision to administer hemostatic agents, however, is most commonly based on clinical assessment. Available hemostatic agents are described in detail below and a summary is provided in Table 2.12 If no response occurs within 12 to 24 hours of administration with the first agent, then an alternative therapy should be considered.
| Table 2. Characteristics of Recombinant Porcine Sequence Factor VIII B-domain-deleted Product12 |
| Drug name (brand) |
Antihemophilic factor (recombinant), porcine sequence (Obizur) |
| Mechanism of action |
Temporarily replaces inhibited endogenous factor VIII |
| Dosing |
200 units/kg every 4-12 h; titrate dose and frequency on the basis of factor VIII response recovery levels and individual clinical response
- Minor-to-moderate bleeds: maintain factor VIII level at 50-100 units/dL or % of normal
- Moderate-to-severe bleeds: maintain factor VIII level at 100-200 units/dL or % of normal for acute bleeds and at 50-100 units/dL or % of normal after acute bleed is controlled, if required
|
| Special populations |
- No dose adjustments for hepatic or renal function
- Efficacy not established in pediatric patients
- No dose adjustments for geriatric patients
|
| Contraindications |
Life-threatening hypersensitivity reactions to antihemophilic factor (recombinant), porcine sequence or its components, which include hamster proteins. Reconstituted product includes: sodium chloride, tris-base, tris-HCl, tri-sodium citrate dehydrate, calcium chloride dehydrate, sucrose, and polysorbate 80. |
| Side effects |
Development of inhibitor antibodies (26% of those who had negative titers at baseline developed antibodies following exposure; 80% of those with positive titers at baseline experienced non-detectable titers following treatment) |
| Monitoring |
- Factor VIII activity level 30 minutes and 3 hours after initial dose and 30 minutes after subsequent doses
- Inhibitory antibodies if plasma factor VIII levels fail to increase as expected or bleeding is not controlled
|
| Storage |
- Store vials in refrigerator and in original package to protect from light
- Reconstituted product expires within 3 hours
|
| Pregnancy/lactation |
- No known studies in pregnant or lactating women
- Pregnancy category C
- Use caution in nursing mothers
|
Replacement therapy: Replacing factor VIII is only effective in patients with low titers (< 5 BU/mL).3 Essentially, replacement of factor VIII is used to overcome the autoantibodies inhibiting the endogenous factor VIII; it is administered with a low starting dose and the dose is slowly increased. The porcine version of factor VIII is very effective in achieving measurable factor VIII levels and hemostasis in AHA.
For decades, a mainstay of treating AHA was the use of plasma–derived porcine factor VIII concentrate (Hyate:C).13 However, because of the risk of viral transmission and allergic reactions, it was withdrawn from the market in 2005. In 2014, a recombinant porcine sequence factor VIII B–domain–deleted product (rpFVIII; Obizur) was introduced.12 This product is produced in a baby hamster kidney cell line and manufactured using viral clearance steps to reduce any viral transmission. Because AHA is a rare disease, only 1 study of the use of rpFVIII in this disease has been conducted. The phase II/III, prospective, open–label study evaluated the efficacy of rpFVIII in 28 AHA patients with acute bleeding episodes.13 rpFVIII was initiated at a dose of 200 units/kg intravenously and then adjusted by the investigator on the basis of clinical status of the patients and by targeting factor VIII levels of greater than 80% during the first 24 hours post–infusion and 50% or greater thereafter. Hemostasis and factor VIII levels increased to greater than 100% within 24 hours in all patients, and 86% of patients had successful control of their initial bleeding episode. No serious safety events, including thrombotic events, allergic reactions, or thrombocytopenia occurred. Safety and efficacy of rpFVIII in AHA was confirmed in a follow–up study, which used lower loading doses (100 units/kg).14
Bypassing agents: Acute bleeding episodes can also be controlled with bypassing agents, which are blood products that are used to bypass factors needed to inhibit clot formation, or agents aimed at increasing circulating factor VIII levels. Bypassing agents used in the first–line treatment of acute bleeding episodes in AHA include recombinant factor VIIa (rFVIIa; NovoSeven) and activated prothrombin complex concentrate (aPCC; FEIBA).2 Although comparative trials have not been conducted, these 2 products are generally considered to have similar efficacy.
aPCC is a plasma–derived concentrate containing active clotting factors that has undergone dry heat vapor treatment to inactivate any viral contaminates.2 The recommended dose of aPCC is 50 to 100 units/kg administered intravenously every 8 to 12 hours, with a total dose not exceeding 200 units/24–
hour period. Duration of treatment is based on clinician’s determination of when bleeding has subsided. aPCC is associated with an 86% to 93.3% efficacy.2,3 Severe bleeding typically resolves with a median of 10 infusions and moderate bleeding resolves with 6 infusions.15 Overall, aPCC is tolerated well. In large doses, a rapid rise in the inhibitor titer can occur because the product may contain some factor VIII. Viral transmission is a theoretical concern because the product is human derived; however, no documented cases of viral transmission have been reported. The most common side effect with bypassing agents is thrombosis. The rate of thrombotic events (both arterial and venous) are similar with both bypassing agest, as reported by the EACH2 registry (a multicenter, pan–European database): 2.9% with rFVIIa and 4.8% with aPCC.16 This incidence is higher than that for patients with congenital hemophilia, although this difference is not unexpected, since AHA patients tend to be older and have additional cardiovascular risk factors.2
rFVIIa binds to the surface of activated platelets, supporting thrombin generation, and bypasses the need for factor VIII.2 Initially, rFVIIa was developed for patients with congenital hemophilia, but it has shown efficacy in AHA, as well. The recommended dose is 90 to 120 mcg/kg by intravenous bolus every 2 to 3 hours; it should be administered until bleeding stops.8 Doses may be increased to 120 to 270 mcg/kg if no response is observed after 2 doses. However, patients who do not respond within 24 hours are not likely to respond at all.17 Major bleeding episodes typically require several days of treatment, but minor episodes only require 2 to 3 doses. The efficacy of rFVIIa has been reported to be 95%, with a median dose of 90 mcg/kg (range, 60–160 mcg/kg). The number of infusions and length of therapy, however, are much more variable. The average number of infusions ranges from 1 to 33, with durations of 1 to 7 days.18 EACH2 registry data report a similar efficacy between both agents (aPCC, 93%; rFVIIa, 92%).3,16 Overall, rFVIIa is well tolerated and, because of the recombinant production process, does not carry the potential to transmit human pathogens.
Other hemostatic approaches: Other agents that are sometimes used to control acute bleeding episodes in other bleeding disorders may be considered in AHA when hemostatic and bypassing agents have failed. Desmopressin (DDAVP) is only useful for treating minor bleeds and is effective only in patients with low titers (< 2 BU/mL) and measureable factor VIII levels (> 5 IU/dL).3 Furthermore, tachyphylaxis usually occurs following 3 or more consecutive doses. Overall, DDAVP is well tolerated in most patients, but it should be used cautiously in the elderly because the potential for adverse effects, such as hyponatremia, water intoxication, and subsequent seizures, is high. Tranexamic acid in combination with aPCC or rFVIIa is another potential option. This combination normalizes clot stability. Solutions of tranexamic acid can be applied topically to combat minor skin or oromucosal bleeding and oral tablets may be taken to treat minor systemic bleeding.
Inhibitor eradication
Long–term eradication of the autoantibodies (i.e., the inhibitor) is the only way to restore normal hemostasis and minimize the risk of bleeding.17 Approximately 25% of patients will undergo spontaneous remission after several months, but, at the time of hemostatic inhibition, bleeding–related mortality is high.3,19 Therefore, it is recommended that all adult AHA patients receive inhibitor eradication therapy. The role in pediatric patients is not well established.3 The mainstay of inhibitor eradication is immunosuppression in AHA. Available options are described in Table 3.3 Data supporting the use of immunosuppression are derived from observational studies and registry data comparing the therapies.
| Table 3. Inhibitor Eradication Therapies3 |
| Therapy |
Dose |
Common side effects |
Notes |
| Corticosteroids |
Prednisone 1 mg/kg PO daily or dexamethasone 40 mg PO daily x 4-7 days |
- Nausea/peptic ulcer disease
- Hyperglycemia
- Infection
- Insomnia
- Psychologic effects
|
- Unlikely to be effective in less than 3 weeks in patients with factor VII level < 1 IU/L or inhibitor level > 20 BU/mL
- Periodically monitor glucose levels (monitor frequently in patients with diabetes)
|
| Corticosteroids + cyclophosphamide |
Prednisone 1 mg/kg PO daily or dexamethasone 40 mg PO daily x 4-7 days
+
Cyclophosphamide 1-2 mg/kg PO daily (alternative regimen: 5 mg/kg IV every 3-4 weeks) |
- Nausea/peptic ulcer disease
- Hyperglycemia
- Infection
- Insomnia
- Psychologic effects
- Myelosuppression
- Alopecia or hair thinning
- Nausea
- Diarrhea
- Mucositis
|
- May have faster response rate than corticosteroids alone
- Periodically monitor CBC, kidney and liver function, glucose levels (monitor glucose frequently in patients with diabetes)
- Cyclophosphamide is a hazardous drug, so it requires safe handling by pharmacists, patients, and caregivers
- Provide appropriate education for patients to understand safe handling and appropriate dosing
- Encourage patients to drink 8 to 10 ounces of fluid each day and to empty bladder frequently (to avoid hemorrhagic cystitis)
- Prednisone should be taken with food
- Cyclophosphamide may be taken with or without food, but food may minimize nausea
|
| Corticosteroids + rituximab |
Prednisone 1 mg/kg PO daily or dexamethasone 40 mg PO daily x 4-7 days
+
Rituximab 375 mg/m2 IV weekly x 4 doses (alternative regimen: 100 mg IV weekly x 4 doses) |
- Nausea/peptic ulcer disease
- Hyperglycemia
- Infection
- Insomnia
- Psychologic effects
- Infusion-related reactions
- Myelosuppression
- Fatigue
|
- Not recommended as initial monotherapy unless other options are contraindicated
- Monitor CBC weekly to monthly
- Monitor blood pressure and vital signs during infusion
- Monitor for infusion-related reactions during therapy
- Monitor glucose levels periodically (monitor frequently in patients with diabetes)
- Screen all patients for hepatitis B infection before therapy and those who are carriers or with current/recovered infection; monitor for reactivation during and for 2 years following therapy
- Watch for signs of progressive multifocal leukoencephalopathy and obtain MRI immediately if signs/symptoms present
|
| Abbreviations: CBC, complete blood count; IV, intravenously; MRI, magnetic resonance image; PO, by mouth |
Overall, the combination of corticosteroids with a cytotoxic agent like cyclophosphamide or rituximab is more effective in achieving complete remission than corticosteroids alone. Registry data show that the combination of cyclophosphamide and corticosteroids results in a complete remission rate of 80% within a median of 40 days (interquartile range [IQR], 18–80 days).20 The combination of rituximab and corticosteroids results in a complete remission rate of 61% within a median of 64 days (IQR, 82–206 days), and corticosteroids alone results in a complete remission rate of 58% within a median of 32 days (IQR, 15–51 days); however, these 2 treatments have not been prospectively compared to one another. Additionally, the time to negative inhibitor and factor VIII levels lower than 70 IU/dL is shorter with the combination therapy of cyclophosphamide plus corticosteroids compared with corticosteroid therapy alone (hazard ratio, 2.11; confidence interval, 1.38–3.21; P < 0.001). Interestingly, no difference in the number of patients alive and inhibitor free at 1 year was observed between the 2 therapies. Based on these complete remission rates reported with this registry data, the combination of cyclophosphamide and corticosteroids is the preferred first–line treatment, if patients are able to tolerate the high rate of adverse effects.3
The only prospective study of immunosuppressive therapy that has been conducted is the Acquired Hemophilia Working Group of the German, Austrian, and Swiss Thrombosis and Hemostasis Society (GTH–AH) study.21 A total of 102 patients were enrolled in the study. Initially, patients received corticosteroids (weight–based dosing) for 3 weeks. If remission was not achieved, cyclophosphamide (weight–based dosing) was added. If patients still did not achieve remission within 4 to 6 weeks, then rituximab (375 mg/m2 weekly) was added. Partial remission (defined as factor VIII levels > 50 IU/dL, no active bleeding, and no hemostatic drugs for at least 24 hours) was achieved in 83% of patients. Complete remission—defined by the same criteria as partial remission in addition to a negative inhibitor test, prednisone dose tapered to less than 15 mg daily, and other immunosuppressive therapy discontinued—was achieved in 61% of patients. The median time to partial remission was 31 days (IQR, 19–51 days) and the median time to complete remission was 79 days (IQR, 48–102 days).
If the approach defined in the GTH–AH study is ineffective, the use of other therapies may be warranted.3 However, very limited data are available that support the use of other immunosuppressive therapies, such as calcineurin inhibitors, mycophenolate mofetil, and combination immunosuppressive therapy. The use of immunoglobulin has shown no additional benefit and should not be routinely administered.
MONITORING THERAPY
To optimize outcomes and mitigate adverse events, patient monitoring is critical during any type of therapy for AHA. Both hemostatic therapy and immunosuppressive therapy require clinical observation of the patient’s signs and symptoms, including physical examination and laboratory testing.
Hemostatic therapy
Monitoring response following hemostatic therapy is based on clinical assessment, hemoglobin levels, and imaging studies.3 The expectation is that, following hemostatic therapy, the patient’s bleeding will subside as observed through physical examination and, if needed for internal bleeding sites, imaging studies. The hemoglobin should rise following hemostatic therapy.
Laboratory tests have yet to be standardized for monitoring the response to bypassing agents; however, laboratory tests can be used following therapy with factor VIII agents.3 With rpFVIII, routine factor VIII levels using 1–stage coagulation assays should be conducted. Typically, the assays should be obtained in real time, rather than sent to an outside laboratory, so that clinicians can respond with changes in therapy, if needed. Realistically, a turnaround time of 1 to 2 hours is needed to successfully manage these patients.
In the study evaluating rpFVIII, during the first 24 hours, factor VIII activity was measured within 10 to 20 minutes pre–infusion and post–infusion and repeated every 2 to 3 hours with a target level of greater than 80% for significant bleeds (i.e., severe mucosal, intracranial, retro– or intra–abdominal, genitourinary, neck, traumatic, and post–operative bleeds) and greater than 50% for other bleeds.13 After 24 hours, the dose was titrated to target post–infusion factor VIII levels of 50% or greater for all bleeds. Complete efficacy was measured by cessation of bleeding or related symptoms (e.g., decreased swelling, improved range of motion, tenderness resolved), clinical control as assessed by the provider, and factor VIII activity levels of 50% or greater.13 Partial efficacy was measured by reduced bleeding, clinical stabilization or improvement of clinical status, and/or alternative reason for bleeding and factor VIII activity levels of 20% or greater. The manufacturer of rpFVIII recommends monitoring factor VIII activity 30 minutes and 3 hours after the initial dose and 30 minutes after all subsequent doses.12 Additionally, it is important to monitor the development of inhibitory antibodies to the product itself, particularly if the expected result is not obtained.
Monitoring aPTT can also be useful, since it normalizes when replacement results in factor VIII levels greater than 30% to 50%.3 However, aPTT does not predict when factor VIII normalizes. Thus, aPTT is not considered a good surrogate marker to guide treatment: factor VIII levels are preferred and are the standard of care. Further, no specific factors, such as residual factor VIII activity levels or inhibitor titers, have been shown to predict response to hemostatic therapy.
Inhibitor eradication
Data supporting appropriate monitoring in patients receiving immunosuppression for eradication of autoantibodies are lacking. A consensus panel recommends monitoring factor VIII activity levels and inhibitor titers weekly until factor VIII activity levels normalize and undetectable amounts of inhibitors are present.3 The median time to response of a factor VIII activity level restored to 50 IU/dL or more is 5 weeks; however, those with factor VIII activity levels less than 1 IU/dL at baseline often require significantly longer times on therapy before response is observed. Tapering of corticosteroids should begin as soon as the inhibitor is undetectable and factor VIII activity levels begin to rise. Continued monitoring is required during the tapering period and patients should be educated on the need to return to the clinic for monitoring and evaluation, as well as the need to report new symptoms to the heathcare provider immediately.
THE SPECIALITY PHARMACIST’S ROLE IN AHA
Specialty pharmacists can play a pivotal role in managing patients with AHA. Pharmacists should review patients’ medication lists throughout their courses of AHA treatment to ensure that patients are not using aspirin or non–steroidal anti–inflammatory drugs for pain. They should also ensure that other anticoagulants are not prescribed, unless a hematologist is involved with that decision. Aspirin and anticoagulants may be appropriate in patients at risk for a thrombosis, but these should only be used when FVIII levels are > consistently above 50 IU/dL and the patient is no longer bleeding.3 Pharmacists should also be involved in educating patients and caregivers not only about appropriate storage and handling (particularly of the oral formulation of cyclophosphamide) but also proper administration and common and serious side effects of therapies.22 Pharmacists should educate patients about the need to wear or carry identifying information about the disease, as well as provider contact information, in the event of an emergency and when and where to seek emergency care if a bleeding event happens.3
Drugs used to manage AHA are often in the highest tier for insurance copays, which means that patients may require financial assistance, and pharmacists can assist with finding support for patients. Additionally, the specialty pharmacist can ensure that there are adequate supplies of therapies in stock, particularly when AHA patients are undergoing planned procedures. Often, this requires pharmacists to be actively involved in justifying the high cost of these drugs for budget purposes and to have procedures in place to obtain hemostatic agents quickly and minimize waste. Pharmacists should be involved in calculating or double–checking the dosing of these agents and ensuring that appropriate monitoring is completed following administration. Pharmacists should follow the laboratory results and monitor for response and the need for subsequent doses. If pharmacists are involved in an interprofessional approach to managing AHA patients, optimal outcomes can be achieved.
REFERENCES
- Escobar MA, Key NS. Hemophilia A and hemophilia B. In: Kaushansky K, Lichtman MA, Prchal JT, et al, eds. Williams Hematology. 9th ed. New York, NY: McGraw–Hill Education; 2016.
- Franchini M, Vaglio S, Maranao G, et al. Acquired hemophilia A: a review of recent data and new therapeutic options. Hematology. 2017;22(9):514–520.
- Kruse–Jarres R, Kempton C, Baudo F, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92(7):695–705.
- Von Depka M. Novoseven: mode of action in acquired haemophilia. Intensive Care Med. 2002;28 Suppl 2:S222–S227.
- Arruda VR, High KA. Chapter 141: Coagulation disorders. In: Kasper DL, Fauci AS, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw–Hill Education; 2016.
- Franchini M, Capra F, Nicolini N, et al. Drug–induced anti–factor VIII antibodies: a systematic review. Med Sci Monit. 2007;13(4):RA55–61.
- Boggio LN, Green D. Acquired hemophilia. Rev Clin Exp Hematol. 2001;5(4):389–404.
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program. 2006;432–437.
- Tiede A, Eisert R, Czwalinna A, et al. Acquired haemophilia caused by non–haemophilic factor VIII gene variants. Ann Hematol. 2010;89(6):607–612.
- Oldenburg J, Zeitler H, Pavlova A. Genetic markers in acquired haemophilia. Haemophilia. 2010;16 Suppl 3:41–45.
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2–year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109(5):1870–1877.
- Obizur [prescribing information]. Westlake Village, CA: Shire; 2017.
- Kruse–Jarres R, St–Louis J, Greist A, et al. Efficacy and safety of OBI–1, an antihaemophilc factor VIII (recombinant), porcine sequence, in subjects with acquired hemophilia A. Heamophilia. 2015;21(2):162–170.
- Tarantino MD, Cuker A, Hardesty B, et al. Recombinant porcine sequence factor VIII (rpFVIII) for acquired haemophilia A: practical clinical experience of its use in seven patients. Heamophilia. 2017;23(1):25–32.
- Sallah S. Treatment of acquired haemophilia with factor eight inhibitor bypassing activity. Haemophilia. 2004;10(2):169–173.
- Baudo F, Collins P, Huth–Kühne A, et al; EACH2 registry contributors. Management of
bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) registry. Blood. 2012;120(1):39–46.
- Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicenter study. Thomb Haemost. 1997;78(6):1463–1467.
- Sumner MJ, Geldziler BD, Pedersen M, Seremetis S. Treatment of acquired haemophilia with recombinant activated FVII: a critical appraisal. Haemophilia. 2007;13(5):451–461.
- Lottenberg R, Kentro TB, Kitchens CS. Acquired hemophilia. A natural history study of 16 patients with factor VIII inhibitors receiving little or no therapy. Arch Intern Med. 1987;147(6):1077–1081.
- Collins P, Baudo F, Knoebl P, et al; EACH2 registry collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012;120(1):47–55.
- Tiede A, Klamroth R, Scharf RE, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH–AH 01/2010 study. Blood. 2015;125(7):1091–1097.
- Neuss MN, Gilmore TR, Belderson KM, et al. 2016 updated American Society of Clinical Oncology/Oncology Nursing Society Chemotherapy Administration Safety Standards, Including Standards for Pediatric Oncology. Onc Nurs Forum. 2017;44:31–43.
Back Top