
Expired activity
Please go to the PowerPak
homepage and select a course.
Chronic Hyperkalemia: Evolving Options for a Common Problem
INTRODUCTION
Potassium (K+) is the most abundant cation in the body. It is found inside skeletal muscle, liver, and red blood cells, with concentrations approximating 150 mEq/L. Only 2% of total body K+ is found in the extracellular space, mainly in serum and interstitial fluid, at concentrations of 3.5-5.0 mEq/L. Potassium is a dynamic cation, transported intracellularly through the Na+-K+-ATPase pump, which moves sodium (Na+) out of and K+ into cells at a fixed ratio of 3:2. This specific ratio maintains a concentration gradient across cell membranes that is critical to the function of excitable tissue, such as nerve, muscle, and heart.1 In controlling the electrochemical potential of these cells, potassium plays crucial roles in cardiac, digestive, and muscular function.
Potassium is maintained within a narrow physiologic range, foreshadowing the harmful effects of K+ disorders. Ideally, intake matches excretion. When the body senses elevated serum K+, it works to correct the imbalance in 2 ways2:
- Increased K+ excretion
- Redistribution of K+ intracellularly
Cellular shifts correct imbalances faster than excretion using the Na+-K+-ATPase pump under the influence of hormones and acid-base changes.2
Catecholamines, specifically beta-2-adrenergic agonists, activate the Na+-K+-ATPase pump through phosphorylation, shifting K+ into cells.3 Alpha-adrenergic agonists work differently catecholamines, impairing the body’s ability to buffer high K+ loads, which can lead to hyperkalemia.4 Insulin increases the activity and expression of the Na+-K+-ATPase pump through protein kinase signaling.3 Metabolic alkalosis enhances cellular uptake of K+; metabolic acidosis impairs cellular uptake of K+.
The kidneys are the major sites of K+ excretion, with a minor amount excreted by the gastrointestinal (GI) tract. Consequently, the adaptive mechanisms in response to K+ derangements come mostly from increased renal K+ excretion. Potassium is filtered by the glomerulus and a majority is reabsorbed at the proximal tubule and loop of Henle. Only a small fraction (approximately 10%) reaches the distal tubule and collecting duct. The cortical collecting duct (CCD) is the main site of K+ secretion, and is tightly regulated. The CCD’s lumen consists of 75% principal cells and 25% intercalated cells. Principal cells secrete K+ by first moving the cation intracellularly using the Na+-K+-ATPase pump on the basolateral membrane. Then, they move potassium extracellularly into the lumen using ionic conductance channels (renal outer medullary K+ [ROMK] and big K+ [BK]) on the apical membrane.5 Figure 1 depicts K+ secretion in the CCD in detail. As serum K+ normalizes, negative feedback controls engage.6
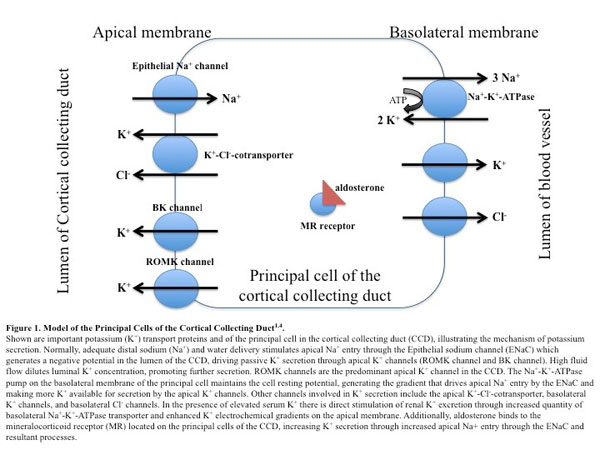 |
Potassium excretion through the GI tract happens passively through a paracellular route and actively through secretion by gut BK channels.6 When renal function is impaired and urinary K+ excretion is markedly decreased (as in severe chronic kidney disease [CKD]), colonic K+ excretion is increased through upregulation of gut BK channels and changes in regulatory signals to promote K+ secretion.6
Another feedback control mechanism occurs through mineralocorticoid stimulation. Mineralocorticoid activity and delivery of Na+ and water to the distal tubule regulates renal K+ secretion. Aldosterone is the major mineralocorticoid in humans, secreted from the adrenal glands in response to several stimuli including elevated serum K+ levels (Figure 1). When elevated serum K+ stimulates aldosterone secretion, it inhibits renin (and therefore angiotensin II). Normally, angiotensin II prevents excessive kaliuresis by inhibiting ROMK channels.4 However in this situation, angiotensin II cannot influence K+ secretion and aldosterone alone manipulates K+.
To prevent aldosterone-driven K+ secretion from causing significant Na+ reabsorption and clinical consequences, other mechanisms of K+ secretion dominate. For example, hyperkalemia stimulates natriuresis, thereby increasing distal Na+ and water delivery.4 This increased tubular flow can stimulate sufficient K+ secretion without aldosterone.4 Additionally, K+ excretion by the ROMK channel can be enhanced or reduced according to serum K+ levels, as a way to regulate K+ excretion independent of Na+ reabsorption.4 Conversely, aldosterone can stimulate Na+ reabsorption using the thiazide-sensitive Na+-Cl--cotransporter, which maintains electroneutrality to avoid stimulating K+ secretion.4 Referred to as the aldosterone paradox, these mechanisms can overcome aldosterone's interdependent effects on Na+ and K+ regulation and allow each electrolyte to be manipulated separately.7
Researchers have discovered an independent feed-forward homeostatic mechanism that drives K+ secretion during meals; splanchnic and brain sensors increase expression of the ROMK channels in the CCD to result in kaliuresis.1 They have also discovered a circadian clock in the brain, controlled by the ambient light-dark cycle, that regulates K+ homeostasis based on food intake.1
EPIDEMIOLOGY OF HYPERKALEMIA
Hyperkalemia is defined as a serum concentration greater than 5.0 mEq/L. The incidence of hyperkalemia in the general population is low, around 2% to 3%,8,9 but the condition occurs in up to 10% of hospitalized patients.10 Development of hyperkalemia can be acute or chronic, depending on the time of onset, which is closely related to the underlying etiology(s). Its severity is determined based on acuity, absolute serum K+ values, and the presence of symptoms.
Hyperkalemia can be caused by pathophysiologic processes in certain disease states, including diabetes (DM), cardiovascular disease (CVD), heart failure (HF), and CKD. In patients with these comorbidities, the reported incidence of hyperkalemia is as high as 50%.8 As glomerular filtration rate (GFR) declines in CKD, the kidneys lose their ability to maintain serum K+ within normal limits. Early on, the remaining nephrons are capable of maintaining normokalemia, but with progression to CKD stage 3 and beyond, this adaptive process eventually fails. Decreased GFR and tubulointerstitial damage in acute kidney injury can interfere with renal K+ excretion. Relative insulin deficiency and decreased mineralocorticoid activity leads to hyperkalemia in DM. Renal hypoperfusion in HF stimulates aldosterone secretion, resulting in decreased distal Na+ delivery and subsequent decreased K+ secretion.
Medications that are part of guideline-recommended therapy for patients with these diseases often cause hyperkalemia. Agents that inhibit the renin-angiotensin-aldosterone system (RAASi) — including angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), aldosterone receptor antagonists (ARAs), and direct renin inhibitors — are common causes of iatrogenic hyperkalemia. Hyperkalemia has become more prevalent as RAASi therapy has proven beneficial in many diseases and is used more commonly. One study of patients 66 years of age or older showed a spike in dual ACEI/ARA prescribing after the publication of RALES, from 34 per 1,000 patients in 1994 to 149 per 1,000 patients in 2001(P < 0.001).11 These investigators also found that publication of RALES was associated with 1,485 additional hyperkalemia-related hospitalizations and 171 additional in-hospital deaths during a 1-year period.11
In patients without risk factors for hyperkalemia, the incidence with RAASi monotherapy is less than 2%.12 That incidence increases to 5% to10% in patients with HF or CKD, but the adverse effect in these patients may not be clinically important.12 Clinical trials tend to have run-in periods to exclude patients who cannot tolerate the medication adverse effects such as hyperkalemia. Clinical trials also provide extensive laboratory surveillance that may be unavailable in clinical practice. Combination RAASi increases the risk of hyperkalemia to 1.2% to 15.6%.13,1
Predictors/Causes of Hyperkalemia
Most causes of hyperkalemia are transient, usually leading to acute episodes. Chronic hyperkalemia is usually related to impaired renal excretion. It is important to assess the cause(s) of hyperkalemia and make note of its duration, which may dictate treatment. This article focuses on causes and treatment of chronic hyperkalemia, with a brief mention of acute causes that can exacerbate the overall disorder.
One generally transient cause of hyperkalemia is excessive dietary potassium intake. Dietary recommendations for patients with HF, CKD, or DM tend to emphasize sodium restriction. Patients who switch to salt substitutes with good intentions often do not realize that the substitute is a K+ salt. Additionally, “heart-healthy” diets tend to include foods with high potassium levels, such as fruits, vegetables, and whole grains. In the presence of normal renal and adrenal function, excess potassium ingestion rarely leads to hyperkalemia.3 However, with underlying renal or adrenal impairment, the adaptive mechanisms that increase renal K+ excretion cannot be fully realized, so the body relies more heavily on increased colonic K+ excretion.3 Ultimately colonic adaptive changes are insufficient and hyperkalemia can develop.
Chronic hyperkalemia from impaired renal excretion may involve 1 or more of the following: decreased mineralocorticoid activity, decreased distal Na+ delivery, and CCD dysfunction.2 Considered individually, these pathophysiologic processes generally do not create chronic hyperkalemia, but together their effects become clinically significant. As GFR declines over time, these processes are critical to maintaining adequate K+ secretion, and lead to hyperkalemia when they fail.15 However, these processes are often connected to disease states and medications that can impair GFR in the first place, inhibiting homeostasis.
RAAS activation leads to increased Na+ and water reabsorption and K+ excretion. When any part of this process is inhibited, hyperkalemia can develop. Hyporeninemic hypoaldosteronism is a syndrome that leads to hyperkalemia through decreased mineralocorticoid activity and can be caused by advancing age, diabetic nephropathy, and interstitial renal disease.4,15 Any form of adrenal insufficiency (e.g., Addison’s disease) can also contribute to hyperkalemia. In addition to the various classes of RAASi, nonsteroidal anti-inflammatory agents, beta-blockers, calcineurin inhibitors, and heparin lead to decreased mineralocorticoid activity. Table 1 describes the mechanisms by which selected medications cause hyperkalemia.
| Table 1. Mechanisms of Drug-Induced Hyperkalemia4,15 |
| Mechanism |
Drugs Implicated |
| Drugs that cause a K+ shift from ICF to ECF |
|
| Decrease ability of Na+-K+-ATPase pump to redistribute K+ intracellularly |
Beta-blockers
Calcineurin inhibitors (tacrolimus and cyclosporine)
Digoxin
IV cationic amino acids |
| Increase osmotic load causing cellular efflux of water and K+ |
Glucose
Mannitol
Radiocontrast material |
| Drugs that interfere with K+ excretion |
|
| Inhibit production or release of renin |
Beta-blockers
COX-2 inhibitors
Cyclosporine
Direct renin inhibitors (aliskiren)
NSAIDs |
| Decrease production or secretion of aldosterone |
ACEIs
ARBs
Beta-blockers
Calcineurin inhibitors (tacrolimus and cyclosporine)
COX-2 inhibitors
Heparins
NSAIDs |
| Antagonize the mineralocorticoid receptor in the nephron |
ARAs |
| Block the epithelial Na+ channel in the cortical collecting duct |
Pentamidine
Potassium-sparing diuretics
Trimethoprim |
| Inhibit K+ conductance channels in the cortical collecting duct |
COX-2 inhibitors
Cyclosporine
NSAIDs |
| Drugs that contain high amounts of K+ |
Herbals (alfalfa, dandelion, horsetail, milkweed, nettle)
Licorice
Penicillin G potassium
Potassium supplements
Salt substitutes |
| Abbreviations: ACEIs, angiotensin-converting enzyme inhibitors; ARAs, aldosterone receptor antagonists; ARBs, angiotensin receptor blockers; ECF, extracellular fluid; ICF, intracellular fluid; IV, intravenous; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Potassium secretion is favored in the setting of increased distal delivery of Na+ and increased distal fluid flow.7 When distal Na+ delivery decreases, the nephrons rely on aldosterone-driven K+ secretion to restore normokalemia. To have a clinically significant impact on K+ secretion, urine Na+ concentrations usually have to be below 10 to 20 mEq/day.16 This may occur in patients with advanced HF and severe CKD, in whom renal hypoperfusion decreases distal Na+ delivery and cannot be overcome by other mechanisms of K+ secretion (i.e., increased mineralocorticoid activity).15 In these settings, use of RAASi therapy exacerbates hyperkalemia.
Abnormal functioning of the CCD can also lead to hyperkalemia. Acute and chronic tubulointerstitial renal disease from conditions such as renal transplantation, systemic lupus erythematosus, amyloidosis, and sickle cell disease directly impair K+ secretion at the CCD.15
Any condition that destroys cells in the body, such as tumor lysis syndrome or rhabdomyolysis, will expel intracellular contents and elevate serum K+. Clinicians must rule out pseudohyperkalemia, an artificial increase in serum K+, before initiating treatment for acute or chronic hyperkalemia. Preparation for venipuncture including fist clenching and tourniquet use can increase K+ efflux from skeletal muscle.4 Thrombocytosis, leukocytosis, or erythrocytosis can all cause pseudohyperkalemia secondary to release of intracellular contents, and serum sample contamination from using a particular anticoagulant or delayed processing of a blood sample can falsely elevate K+ levels.4
Consequences of Hyperkalemia
The clinical significance of hyperkalemia depends on various factors such as the degree of increase from baseline and how quickly it develops. Severe hyperkalemia (usually serum K+ exceeding 6.0 mEq/L) predisposes patients to cardiac arrhythmias and sudden cardiac death, yet serum K+ levels correlate poorly with cardiotoxicity.4 Excess extracellular K+ lessens the negative resting potential of cells, which can affect conduction velocity and repolarization rates; subsequent electrocardiographic changes can manifest as bradyarrhythmias, ventricular fibrillation, and asystole in severe cases.17 Hyperkalemia resulting in marked depolarization of muscle cells inactivates Na+ channels, leading to cell inexcitability and muscle weakness.4 Hyperkalemia also decreases urinary acid excretion. The kidneys normally excrete acid in the form of ammonium. Hyperkalemia inhibits renal acid excretion by competing with ammonium for reabsorption in the loop of Henle and as a result impairs acid secretion further down the tubule.4
In patients with end-stage renal disease (ESRD) on hemodialysis (HD), clinicians should always consider the potential consequences of correcting hyperkalemia. Using a dialysate with a K+ concentration lower than the serum level will create a gradient favoring K+ removal.3 However, a potassium-free dialysate can prevent diffusion of bicarbonate into the serum and inhibit correction of metabolic acidosis. Using a glucose-free dialysate may decrease insulin secretion, keeping more K+ in the extracellular space and available for removal during HD.3 Lowering serum K+ during HD may increase systemic vascular resistance and induce rebound hypertension postdialysis.3 For these reasons, clinicians must address the dialysate Na+ concentration. As serum K+ drops during HD, patients may be more prone to cardiac arrhythmias, especially when hyperkalemia is corrected rapidly.3
Hyperkalemia has been linked to increased mortality. In patients hospitalized for acute myocardial infarction, hyperkalemia was associated with higher in-hospital mortality.18 In a large, retrospective analysis in patients with estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2, the mortality-adjusted incidence rate ratio was 1.6 for patients with serum K+ levels of 5.5–5.9 mEq/L but increased to 3.31 when serum K+ levels reached or exceeded 6.0 mEq/L (P < 0.001).19 An evaluation of 2 large observational studies in adults without CVD found that patients with serum K+ concentrations of 5.0 mEq/L or greater had 41% higher mortality risk (95% CI 1.12–1.76) than those with serum K+ concentrations between 4.0 and 4.4 mEq/L, including a 50% (95% CI 1.00–2.26) higher risk for CVD death.9
Hyperkalemia can prevent patients from receiving optimal disease-specific pharmacotherapy, particularly RAASi. Anticipation of this adverse effect can lead clinicians to avoid prescribing medications, order suboptimal doses, or discontinue key medications prematurely. Discontinuation rates of RAASi in clinical trials are low (1% to 5%),11 but rates in real-world practice are likely higher. A meta-analysis found that combination RAASi was associated with a significantly greater incidence of discontinuation due to adverse effects than monotherapy,11 yet in some studies rates of hyperkalemia were similar between groups. Luo et al. found that clinicians frequently responded to hyperkalemia in patients with CKD by discontinuing RAASi therapy, and only 1.1% of patients had resumed RAASi therapy 30 days later.19 This may reveal a divergence between the perception and actual occurrence of hyperkalemia from RAASi or demonstrate that the clinical impact of hyperkalemia from RAASi can be overcome by vigilant monitoring.
Case Study 1
A 53-year-old woman with reduced ejection fraction HF, New York Heart Association class III, and eGFR 54 mL/min/1.73 m2 receives guideline-directed medical therapy, including an ACEI, a beta-blocker, and a loop diuretic. Her electrolytes are within normal limits. Her prescriber adds spironolactone because of continuing HF symptoms. Two weeks later, her serum K+ is 5.8 mEq/L. What is the best plan to treat her hyperkalemia, and what should be done to prevent hyperkalemia from recurrence?
We will return to this case at the end of the article.
TREATMENT OF CHRONIC HYPERKALEMIA
Acute symptomatic hyperkalemia is a clinical emergency, and goals of care are different than for chronic hyperkalemia. In acute hyperkalemia, treatment goals include symptom resolution (especially cardiac arrhythmias) and immediate decrease of serum K+ to within normal limits using therapies such as intravenous calcium (Ca2+) gluconate, insulin, and beta-2 agonists.17
For chronic hyperkalemia that is usually asymptomatic, treatment goals include prevention of generalized symptoms, cardiac dysrhythmias, and death. The aim is to maintain serum K+ levels within normal limits or as close to normal as possible. Identification of modifiable (i.e., diet and medication) and nonmodifiable (i.e., CKD, DM) contributors is important. Discontinuation of hyperkalemia-inducing medications is often the first step; however, doing so may deprive the patient of important clinical benefits of the medication.17 RAASi in particular have proven cardiovascular and renal benefits in patients with CKD, DM, HF, and CVD. Guidelines strongly encourage continuing them.20-22 Several medications shown to treat and prevent hyperkalemia may allow continued use of these life-saving therapies.
Treatment Options
Treatment of chronic hyperkalemia includes diuretics, bicarbonate salts, dietary restriction, potassium binders, and dialysis. As described above, K+ is mainly eliminated by the kidneys. While the GI tract plays a minor role, this route of elimination may become more important in patients who have severe renal impairment. Indeed, both active and passive secretory mechanisms, such as up-regulation of gut BK channels, can increase K+ secretion up to 20 mEq to 80 mEq/d in patients with CKD.6 While these adaptive mechanisms do not generally prevent hyperkalemia singlehandedly, they identify potential therapeutic targets for prevention and treatment of chronic hyperkalemia. This article focuses medications that target the GI tract as a route of K+ elimination for chronic hyperkalemia (Table 2).
| Table 2. Comparison of Potassium Binders |
| |
Sodium Polystyrene Sulfonate23 |
Calcium Polystyrene Sulfonate34 |
Patiromer Sorbitex Calcium36,38-39 |
Sodium Zirconium Cyclosilicate40-42 |
| Type of compound |
Nonabsorbable, organ polymer in a sorbitol base |
Nonabsorbable, organ polymer |
Nonabsorbable, organ polymer and sorbitol complex |
Nonabsorbable, insoluble inorganic crystal |
| Mechanism of action |
Exchanges Na+ for H+ in the stomach, then exchanges H+ for other cations in the large intestine |
Exchanges Ca2+ for K+ and Mg2+ |
Preferably binds K+ in exchange for Ca2+; also binds Mg2+ |
Preferably binds K+ in exchange for Na+ and H+; also binds ammonium |
| Location of K+ binding |
Colon/rectum |
Distal colon predominantly |
Distal colon predominantly |
Entire intestinal tract |
| Formulation |
Suspension in sorbitol or dissolvable powder |
Suspension in sorbitol or dissolvable powder |
Powder for oral suspension |
Oral suspension |
| Onset of action |
2–6 hours |
NA |
7–48 hours |
1–6 hours |
| Dosing |
Oral: 15 g one to 4 times daily
Rectal: 30-50 g every 6 hours |
Oral: 15 g 3 to 4 times daily
Rectal: 30 g daily |
Oral: 8.4-25.2 g once daily with food |
Oral: 1.25-10 g three times daily x 48 hours then 1.25-15 g once daily* |
| Place in therapy |
Commonly used for acute hyperkalemia, although evidence is questionable. Minimal data for chronic treatment but appears to be safe and effective for the shorter term. Notable adverse effects: GI injuries, constipation, diarrhea, hypernatremia. |
Approved outside the United States. Effective long term for mild-to-moderate chronic hyperkalemia. Notable adverse effects: GI injuries, constipation, hypercalcemia, hypomagnesemia. |
Effective long term for chronic hyperkalemia, allowing patients to stay on RAASi therapy. Not studied in acute setting or in severe, symptomatic hyperkalemia. Notable adverse effects: constipation, hypokalemia, hypomagnesemia. |
Not FDA approved. If approved, may be suitable for both acute and chronic treatment due to quick onset of action and ability to maintain normokalemia longer term. Notable adverse effects: GI disorders, edema. |
*Sodium zirconium cyclosilicate not FDA approved, dosing based on trial data.
Abbreviations: FDA, Food and Drug Administration; GI, gastrointestinal; NA, not available; RAASi, renin-angiotensin-aldosterone system inhibitor. |
Reversible Etiologies
The first step in managing chronic hyperkalemia is eliminating correctable causes. A review of the patient’s diet is important, and education about potassium-restricted diets should be considered for patients with CKD. Many medications, supplements, and herbals cause hyperkalemia to varying degrees (Table 1) and the effects are always additive. Depending on the acuity of hyperkalemia, medications may be dose-reduced, temporarily withheld, or permanently discontinued. A thorough medication review and consideration of each medication's risk-benefit profile is imperative. In patients with ESRD, clinicians should consider the possibility of inadequate dialysis.
Sodium Polystyrene Sulfonate in Sorbitol
The U.S. Food and Drug Administration (FDA) approved sodium polystyrene sulfonate (SPS), a cation-exchange resin, in 1958 for the treatment of hyperkalemia. SPS exchanges Na+ for Ca2+, ammonia, magnesium (Mg2+), and K+ in the GI tract.23 Because of its nonspecific binding, a 30-g dose binds and causes excretion of only about 10 mEq of K+.23 Under acidic conditions, the sulfonate group is bound by H+ and cannot bind K+, making it most effective in the slightly acidic to neutral colon/rectum.24 SPS expands in water, slowing its movement through the GI tract.
SPS's clinical effectiveness is an area of great debate. The drug product was approved by FDA before passage of the 1962 Kefauver-Harris Amendment, which requires that pharmaceutical companies provide clinical proof of drug safety and effectiveness. Many critics maintain that FDA would probably not approve SPS today. Two studies published in 1961 provided evidentiary support for its continued approval after the 1962 amendment. Scherr et al. tested the SPS powder suspended in water in patients with hyperkalemia and acute or chronic renal failure. The study investigators administered SPS orally to 22 patients, rectally to 8 patients, and chronically to 2 patients. Mean decrease in serum K+ was 1 mEq/L and 0.8 mEq/L within 24 hours for the oral and rectal doses, respectively. Constipation occurred but was “easily controlled” by enemas and cathartics.25
Flinn et al. tested a new formulation of SPS in sorbitol in 10 hospitalized patients with severe oliguria. Treatment consisted of SPS administered as 15-g doses 4 times daily and 70% sorbitol syrup administered every 2 hours.26 Three patients received sorbitol without SPS. Serum K+ levels decreased over a period of 5 days in all patients (no statistical analysis). The decrease in serum K+ concentrations was greater for patients who received sorbitol only. Patients in the SPS and sorbitol groups experienced a rise in serum Na+ that was not seen with the sorbitol-only group, and study investigators suggested that the risk of hypernatremia and edema could be prevented by “drastic restriction of water."26 Of note, the SPS product’s sodium content is 100 mg (4.1 mEq) per gram, and product labeling advises against its use in patients who cannot tolerate even small increases in sodium loads.23 In both studies, SPS's K+ lowering effect was marginal, yet SPS continues to be used frequently for the acute treatment of hyperkalemia.10
SPS's hypokalemic effects may be driven by the cathartic (sorbitol) rather than the resin itself. This was first suggested by the Flinn et al. study26 and supported by a later study that showed that the addition of SPS to a laxative did not significantly increase fecal K+ output compared with various laxatives alone (P < 0.02 for phenolphthalein/docusate).27 In 1982, FDA approved a premixed suspension of SPS in sorbitol, and it quickly became the main SPS product purchased by hospitals and pharmacies.28 Over the years, numerous adverse event reports detailed cases of serious GI injuries including intestinal necrosis from SPS with and without sorbitol. Most cases occurred with SPS in 70% sorbitol, with enema administration, or postoperatively.3,4 Though the incidence of this complication appears low, it can be fatal in up to 33% of affected patients.29
In response to these reports, FDA changed SPS's labeling to state that coadministration with sorbitol is not recommended.23 This labeling change did not apply to the premixed formulations containing 33% sorbitol, which is the product stocked by most U.S. pharmacies and hospitals.
While more commonly used in the acute care setting, SPS is also prescribed for treatment and prevention of chronic hyperkalemia with minimal supporting evidence because of the lack of other options. One small (n = 14), retrospective study evaluated long-term use of SPS without sorbitol in patients with CKD and CVD who were being treated with RAASi.30 During the median follow-up of 14.5 months, serum K+ fell from 6.4 mEq/L to 4.6 mEq/L and remained within normal limits.30
More recently, a small randomized, placebo-controlled clinical trial evaluated SPS's efficacy in patients with CKD (eGFR < 40 mL/min/1.73 m2) and asymptomatic hyperkalemia (serum K+ 5.0 to 5.9 mEq/L).31 Thirty-three patients were randomized to a fixed dose of SPS 30 g without sorbitol or placebo orally once daily for 7 days. A majority of the patients were on RAASi. Absolute serum K+ levels were significantly decreased by SPS compared with placebo (-1.04 mEq/L; P < 0.001). By the end of 7 days, 11 of the 15 (73%) patients in the SPS group and 6 of the 16 (38%) patients in the placebo group achieved normokalemia (P = 0.07). Intestinal necrosis did not occur, which is not unexpected given the small sample size and use of SPS without sorbitol.31
Prompted by the safety evaluations of another potassium binder, patiromer, FDA required the SPS manufacturer to conduct studies evaluating the product’s potential to bind to other orally administered medications.32 A subsequent in vitro study found that SPS significantly decreased the absorption of 6 medications (amlodipine, metoprolol, amoxicillin, furosemide, phenytoin, and warfarin). FDA has since issued a communication recommending that patients separate doses of all commercially available SPS products and orally administered prescription and over-the-counter medications by at least 3 hours.33
Calcium Polystyrene Sulfonate
Calcium polystyrene sulfonate (CPS; Resonium Calcium and others) is a cation-exchange resin currently approved in many countries outside the United States. It exchanges Ca2+ for K+ and Mg2+ and works predominantly in the distal colon. CPS is indicated in patients with hyperkalemia associated with anuria or severe oliguria and can also be used in patients on dialysis.34
Canadian prescribing information recommends medication initiation when serum K+ levels are above 6.0 mEq/L and discontinuation when serum K+ concentration is at or below 5.0 mEq/L. Clinicians should avoid using CPS with sorbitol because of case reports of intestinal necrosis.34
A recent study evaluated CPS's long-term efficacy for mild (serum K+ >5.0 mEq/L) hyperkalemia in patients with CKD.35 Most patients had diabetic nephropathy and 62.8% were on RAASi. Normokalemia was achieved in 66.7% to 86.8% of patients in a dose-dependent manner, over a mean treatment duration of 5.6 ± 8.7 months. Of note, patients on RAASi were significantly less responsive to CPS than nonusers (74% vs 86%; P <0.05). Constipation occurred in 8% of patients.35
Patiromer
Patiromer is a cation-exchange resin approved by FDA in 2015 for treatment of hyperkalemia. Unlike SPS, it absorbs water minimally, does not need a laxative to reach the colon, has a higher K+ binding capacity, and uses Ca2+ rather than Na+ as the exchange cation. The product contains sorbitol to increase stability of the drug, with amounts 5-fold to 10-fold lower than the amount coformulated with SPS.6
Two major clinical trials were instrumental in patiromer's FDA approval. The OPAL-HK study was a phase 3, multicenter trial that evaluated patiromer's efficacy and safety in adults with CKD on stable RAASi doses who were hyperkalemic (serum K+ 5.1 to 6.5 mEq/L).36 The study had 2 phases: a 4-week initial treatment phase and an 8-week placebo-controlled, randomized withdrawal phase. During the initial treatment phase, patients were given patiromer 4.2 or 8.4 grams twice daily based on hyperkalemia severity. The mean change in serum K+ concentration at the end of 4 weeks was –1.01 ± 0.03 mEq/L (P < 0.001). Higher baseline serum K+ levels were associated with greater serum K+ lowering. By the end of phase 1, 76% of patients had reached the target serum K+ concentration of 3.8 to 5.0 mEq/L.36
In the second part of the study, patients with a baseline serum K+ concentration exceeding 5.5 mEq/L before the start of phase 1 who also met the target serum K+ concentration after the 4-week treatment were assigned 1:1 to continue patiromer (at the same dose) or switch to placebo. In the placebo group, the estimated median increase in serum K+ concentration after 4 weeks was 0.72 mEq/L. Patients in the patiromer group experienced no K+ increase. Sixty percent of patients in the placebo group versus 15% in the patiromer group had recurrent hyperkalemia (serum K+ ≥5.5 mEq/L) through week 8 of the withdrawal phase. At the end of the withdrawal phase, clinicians discontinued RAASi in 56% of placebo-treated, but discontinued RAASi in only 6% in the patiromer group. During the total 12-week study, the most common adverse effect was constipation (11%) and 3% of patients experienced hypokalemia.36 A post hoc analysis of OPAL-HK demonstrated that patiromer also reduces aldosterone levels and blood pressure.37
The AMETHYST-DN study was a phase 2, multicenter, dose-ranging, randomized trial, It evaluated the efficacy and safety of patiromer in patients with hyperkalemia and diabetic nephropathy.38 The 4-week, run-in phase enrolled patients with uncontrolled blood pressure treated with 1 RAASi who did not have hyperkalemia and aimed to optimize their RAASi dose and/or add a second RAASi to their treatment. Patients who developed hyperkalemia during the run-in phase or were hyperkalemic at baseline (serum K+ 5.0 to <6.0 mEq/L) were included in the 8-week treatment phase and 44-week maintenance phase.38
Patiromer dosing in the treatment phase was based on baseline serum K+ and titrated to achieve and maintain a goal serum K+ level of 5.0 mEq/L or below. Significant reductions in serum K+ were seen as early as 48 hours after patiromer initiation. Normokalemia was achieved and maintained during the entire 52 weeks in 77% to 95% of patients, depending on the severity of baseline hyperkalemia. The most frequently reported treatment-related adverse events were hypomagnesemia (7.2%), constipation (4.6%), and diarrhea (2.7%). Patients experienced blood pressure reductions as early as 3 days after treatment and maintained these throughout the study, possibly because contolling potassium allowed the investigators to continue RAASi or increase doses.38
In clinical trials, patiromer was shown to treat hyperkalemia and maintain normal serum K+ in patients at high risk for chronic hyperkalemia. Patiromer should not be used as emergency treatment for hyperkalemia because of its delayed onset of action.39 Patients should take patiromir at least 3 hours before or after all other oral medications to avoid drug binding and decreased effectiveness. Clinicians should monitor for changes in serum K+, Mg2+, and Ca2+, although the clinical significance of Ca2+ release from patiromer has yet to be elucidated.39
Investigational Agent
Sodium zirconium cyclosilicate (ZS-9) is a cation-exchange resin currently awaiting FDA approval. ZS-9 exchanges Na+ and H+ for K+, is highly selective for K+, and works throughout the entire GI tract.40 Two phase 3, placebo-controlled trials have evaluated its efficacy and safety.
The HARMONIZE trial enrolled ambulatory patients with hyperkalemia to receive ZS-9 (powder mixed with water) 10 g three times daily for 48 hours.41 Patients who achieved normokalemia at 48 hours were randomized to 1 of 3 once-daily treatment groups or placebo for 28 days. A majority of patients had CKD and DM, and 70% of patients were on RAASi. A decrease in serum K+ concentration of 0.2 mEq/L (95% CI, –0.3 to –0.2) was seen 1 hour after the first dose; median time to normokalemia was 2.2 hours. Serum K+ levels normalized in 98% of patients by 48 hours. Between 71% and 85% of patients in the active treatment groups remained in normokalemia at 28 days. In the placebo group, 41% maintained normokalemia. Adverse effects were dose-dependent, with the most common being edema (14.3%) and hypokalemia (10.7%).41
Packham et al. conducted a similar dose-finding study in patients with hyperkalemia (serum K+ 5.0 to 6.5 mEq/L) that included a thrice daily, placebo-controlled initial phase (days 1and 2) and once-daily, placebo-controlled maintenance phase (days 3–15).42 All patients in the active treatment groups reached normokalemia within 48 hours on average, while patients who received placebo experienced little to no change. All doses were significantly better than placebo at maintaining normokalemia (P = 0.008 and P <0.001 for the 5-g and 10-g doses, respectively).42
In combining results from both studies, the median time to serum K+ level below 6.0 mEq/L was 1.07 hours and to serum K+ level below 5.5 mEq/L was 4 hours.43 It has been postulated that ZS-9's onset of action is faster than other K+ binders' because of its ability to bind K+ throughout the GI tract.
PUTTING IT ALL TOGETHER – CASE STUDIES
Follow-up: Case Study 1
Review of treatment options for chronic hyperkalemia allows more careful consideration of the patient presented in case study 1. While the patient was already on 2 medications (ACEI and beta-blocker) that can cause hyperkalemia, her serum K+ level was within normal limits. This is likely because of the coadministration of a loop diuretic and her only moderately impaired renal function. Her underlying HF and CKD, which both elevate risk of hyperkalemia, make starting RAASi therapy a very delicate situation. Addition of spironolactone was the likely cause of her hyperkalemia. In HF, ARAs provide significant cardiovascular and renal protection, even when added to an ACEI or ARB, and the ACEI provides renoprotection in CKD.20,22 Therefore, attempts should be made to continue dual RAASi therapy.
A serum K+ concentration of 5.8 mEq/L is considered moderately elevated; the patient’s health care team should assess her for symptoms, consider evaluating her for electrocardiographic changes, and initiate acute therapy. They should also hold the spironolactone until serum K+ level is below 5.0 mEq/L.22 Once the acute hyperkalemia has resolved, the team needs to take actions to prevent recurrence. Spironolactone should be resumed with a maximum dose of 25 mg daily when used with an ACEI or ARB.15
Alternatively, guidelines recommend 12.5 mg or 25 mg doses given every other day.22 Depending on the patient’s volume status, the dose of loop diuretic may be cautiously increased. As metabolic acidosis is a common complication of CKD, correction with sodium bicarbonate is recommended if acid–base disorder is present.20
Pharmacists should advise the patient not to take over-the-counter and herbal products that can produce hyperkalemia. Close monitoring of her renal function and serum K+ levels, including assessments within 1 week of starting or following RAASi dose escalation is recommended.22 The health care team should also assess this patient's diet and educate her about dietary potassium restrictions. If hyperkalemia is still present or hyperkalemia is anticipated, the team can consider starting a potassium binder (Table 2). All available resins have been shown to be more effective than placebo, but few trials have compared the efficacy between medications. In this case, choosing patiromer or, outside the United States, a calcium-based binder such as CPS may be the best option for avoiding the potential for Na+ absorption and volume overload with sodium-based binders. Patiromer has shown efficacy in patients with HF and reduces blood pressure and aldosterone levels, which may make it the most appropriate binder for this patient.38,44
Case Study 2
A 72-year-old man with stage 3 CKD and a 12-year history of type 2 DM (glycated hemoglobin 12.3%) is treated with guideline-directed pharmacotherapy, including an ACEI. Over the past few months, his eGFR has declined and his serum K+ has increased (now 5.4 mEq/L). He is mildly symptomatic with occasional heart palpitations but without electrocardiographic changes. What are options for his treatment?
In this patient, hyperkalemia has developed gradually. When renal function is impaired, the remaining functioning nephrons can adapt and maintain adequate K+ secretion until GFR is severely diminished. However, this patient is prone to hyperkalemia owing to the hyporeninemic hypoaldosteronism associated with DM and older age.15 Insulin deficiency and increased serum glucose associated with DM further contributes to hyperkalemia. As the hyperkalemia is mild and relatively asymptomatic, acute therapies are unnecessary.
The first step in assessing this patient is review of his diet and use of other mediations that can cause hyperkalemia. The ACEI is contributing to his hyperkalemia. Considering the long-term benefits of ACEI therapy for both his CKD and DM, the health care team can attempt to resolve his hyperkalemia without discontinuing RAASi therapy. The risk of hyperkalemia from RAASi therapy is increased with decreasing renal function; the patient’s eGFR and degree of CKD should be quantified. Long-term diuretics and sodium bicarbonate may not be appropriate based on the patient’s volume status and serum bicarbonate. His CKD has not yet progressed to ESRD, which could correct the hyperkalemia.
Potassium binders are an option for this patient. SPS is effective in patients with CKD who are on stable doses of RAASi, although the benefits have been tested only for a short period and in a small group of patients.31 CPS is not indicated for hyperkalemia until serum K+ concentrations exceed 6.0 mEq/L,34 although studies have evaluated its use in mild hyperkalemia.35 Patiromer is effective in patients with diabetic nephropathy for long-term treatment and prevention of hyperkalemia.38 This drug also often allows patients to continue taking RAASi therapy,36 which is the goal for this patient. As it will take 1 to 2 days to see patiromer's full effect, monitoring the patient’s symptoms until then is important in case acute therapy is needed. Regular monitoring and assessment of the patient while on patiromer therapy is imperative.
REFERENCES
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol. 2014;10(6):1050-1060.
- Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis. 2010;56(2):387-393.
- Toto RD. Serum potassium and cardiovascular outcomes: the highs and the lows. Clin J Am Soc Nephrol.2017;12:220-221.
- Beck FK, Rosenthal TC. Prealbumin: a marker for nutritional evaluation. Am Fam Physician. 2002;65(8):1575-1578.
- Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch Eur J Physiol. 2009;458(1):157-168.
- Li L, Harrison SD, Cope MJ, et al. Mechanism of action and pharmacology of patiromer, a nonabsorbed cross-linked polymer that lowers serum potassium concentration in patients with hyperkalemia. J Cardiovasc Pharmacol Ther. 2016;21(5):456-465. doi:10.1177/1074248416629549.
- Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G. Aldosterone paradox: differential regulation of ion transport in distal nephron. 2011;26(2):115-123.
- Kovesdy CP. Management of hyperkalaemia in chronic kidney disease. Nat Rev Nephrol. 2014;10(11):653-662.
- Hughes-Austin JM, Rifkin DE, Beben T, et al. The relation of serum potassium concentration with cardiovascular events and mortality in community-living individuals. Clin J Am Soc Nephrol. 2017;12:245-252.
- Fordjour KN, Walton T, Doran JJ. Management of hyperkalemia in hospitalized patients. Am J Med Sci. 2014;347(2):93-100. doi:10.1097/MAJ.0b013e318279b105.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004;351(6):543-551.
- Weir MR, Rolfe M. Potassium homeostasis and renin-angiotensin-aldosterone system inhibitors. Clin J Am Soc Nephrol. 2010;5(3):531-548.
- Pitt B, Bakris G, Ruilope LM, DiCarlo L, Mukherjee R. Serum potassium and clinical outcomes in the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS). 2008;118(16):1643-1650.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: A quantitative review of data from randomized clinical trials. Arch Intern Med. 2007;167(18):1930-1936.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin–angiotensin–aldosterone system. N Engl J Med. 2004;351(6):585-592.
- DeFronzo, RA. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney Int. 1980;17:118-134.
- Kovesdy CP. Management of hyperkalemia: an update for the internist. Am J Med. 2015;128(12):1281-1287.
- Goyal A, Spertus JA, Gosch K, et al. Serum potassium levels and mortality in acute myocardial infarction. 2012;307(2):157.
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100.
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Int. 2013;2(2):S139–S274.
- American Diabetes Association. Standards of medical care in diabetes. Diabetes Care. 2017;40:S1-S135.
- Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure. 2013;62(16):e147-e239.
- Kayexalate [package insert]. Laval, Quebec: sanofi-aventis Canada, Inc.; 2014.
- Sterns RH, Grieff M, Bernstein PL. Treatment of hyperkalemia: something old, something new. Kidney Int. 2016;89(3):546-554.
- Scherr L, Ogden DA, Mead AW, Spritz N, Rubin AL. Management of hyperkalemia with a cation-exchange resin. N Engl J Med. 1961;264(3):115-119.
- Flinn RB, Merrill JP, Welzant WR. Treatment of the oliguric patient with a new sodium-exchange resin and sorbitol. N Engl J Med. 1961;264(3):111-115.
- Emmett M, Hootkins RE, Fine KD, Ana CAS, Porter JL, Fordtran JS. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. 1995;108:752-760.
- Holbrook AM. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005;165(10):1095. doi:10.1001/archinte.165.10.1095.
- Harel Z, Harel S, Shah PS, Wald R, Perl J, Bell CM. Gastrointestinal adverse events with sodium polystyrene sulfonate (kayexalate) use: a systematic review. Am J Med. 2013;126(3):264.e9-264.e24.
- Chernin G, Gal-Oz A, Ben-Assa E, et al. Secondary prevention of hyperkalemia with sodium polystyrene sulfonate in cardiac and kidney patients on renin-angiotensin-aldosterone system inhibition therapy. Clin Cardiol. 2012;35(1):32-36.
- Lepage L, Dufour AC, Doiron J, et al. Randomized clinical trial of sodium polystyrene sulfonate for the treatment of mild hyperkalemia in CKD. Clin J Am Soc Nephrol. 2015;10:2136-2142.
- Food and Drug Administration. Drug Safety and Availability - FDA Drug Safety Communication: FDA requires drug interaction studies with potassium-lowering drug Kayexalate (sodium polystyrene sulfonate). https://www.fda.gov/Drugs/DrugSafety/ucm468035.htm. Accessed November 1, 2017.
- Food and Drug Administration. Drug Safety and Availability - FDA Drug Safety Communication: FDA recommends separating dosing of potassium-lowering drug sodium polystyrene sulfonate (Kayexalate) from all other oral drugs. https://www.fda.gov/Drugs/DrugSafety/ucm572484.htm. Accessed November 1, 2017.
- Resonium Calcium [package insert]. Laval, Quebec: sanofi-aventis Canada, Inc.;
- Yu MY, Yeo JH, Park JS, Lee CH, Kim GH. Long-term efficacy of oral calcium polystyrene sulfonate for hyperkalemia in CKD patients. PLoS One. 2017;12(3):1-11.
- Weir MR, Bakris GL, Bushinsky DA, et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med. 2015;372(3):211-221.
- Weir MR, Bakris GL, Gross C, et al. Treatment with patiromer decreases aldosterone in patients with chronic kidney disease and hyperkalemia on renin-angiotensin system inhibitors. Kidney Int. 2016;90(3):696-704.
- Bakris GL, Pitt B, Weir MR, et al. Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease. 2015;314(2):151-161.
- Veltassa [package insert]. Redwood City, CA: Relypsa, Inc; 2016.
- Linder KE, Krawczynski MA, Laskey D. Sodium zirconium cyclosilicate (ZS-9): A novel agent for the treatment of hyperkalemia. 2016;36(8):923-933.
- Kosiborod M, Rasmussen HS, Lavin P, et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia. 2014;312(21):2223.
- Packham DK, Rasmussen HS, Lavin PT, et al. Sodium zirconium cyclosilicate in hyperkalemia. N Engl J Med. 2015;372(3):222-231.
- Kosiborod M, Peacock WF, Packham DK. Sodium zirconium cyclosilicate for urgent therapy of severe hyperkalemia. N Engl J Med. 2015;372(16):1577-1578.
- Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J. 2011;32(7):820-828.
Back to Top