
Expired activity
Please go to the PowerPak
homepage and select a course.
Safe and Efficacious Use of Immuno-Oncology Agents: Pharmacist-Driven Strategies to Improve Outcomes (Article)
INTRODUCTION
Immuno-oncology (IO) agents—known as the fourth treatment modality for cancer—have become an important option for the treatment of many different cancer types and common diseases. The original IO therapy was Coley’s toxin, which dates back to 1890 and consisted of killed bacteria that were injected into tumors, showed only limited success.1 The first IO agent approved for use in cancer was interferon-α for hairy cell leukemia in 1986.2 Since then, more than 26 immunotherapies have been approved for treatment of at least 17 types of cancer.2 Although IO agents have been associated with relatively low objective responses, value is achieved in the extension of survival in responding patients.3 This benefit, commonly described as a durable response, is characterized by a plateau near the end of the survival curve and implies that some patients may be cured from their cancers. Improved understanding of the immune system and identification of key signaling pathways have led to intense research into the development of IO treatments. The large investment in the development of new IO therapies is evidenced by the estimated 940 agents in clinical development and 1064 in preclinical development, according to one analysis in September 2017.2 As a result of these efforts, pharmacists will be seeing an increasing number of these valuable therapies being prescribed for patients with many types of diseases. In addition to changing the standard of care for some diseases, IO agents have side effect profiles that differ from traditional chemotherapeutic agents in the type of toxicity and the time to onset. Toxicity can even vary among different IO agents because of their specific mechanisms of action. Being aware of the potential side effects associated with IO agents, when to expect them, and how to manage them is critical for all members of the treatment team.
MODULATING IMMUNE PATHWAYS: IMMUNE CHECKPOINT INHIBITORS
Historically, IO therapies have focused on making tumors visible to the immune system, which led to modest benefit, usually in uncommon cancers. The recent success with checkpoint inhibition has energized IO research into new treatments. T-cells are a potent element in antitumor activity.3 However, there are negative signaling pathways to dampen activated T-cell responses over time, which are thought to be crucial for self-tolerance. These negative signaling pathways are known as checkpoint pathways and, when activated, lead to exhausted T-cells. Research has shown that inhibiting these checkpoint pathways can reinvigorate exhausted T-cells, which promotes anti-tumor activity and can then be used therapeutically.4 Ipilimumab was the first immune checkpoint inhibitor to be approved by the United States Food and Drug Administration (FDA). It was originally associated with an increase in both median survival and long-term durable responses in metastatic melanoma.5 In a meta-analysis of data from 1861 patients with unresectable or metastatic melanoma (both previously treated [n = 1257] and untreated [n = 604]) in 10 prospective and 2 retrospective studies, the median overall survival (OS) was 11.4 months. At 3 years, the survival rate was 22% in all patients, and there was a plateau in pooled Kaplan-Meier data that began at approximately 3 years after the start of treatment with ipilimumab and extended through 10 years of follow up.5 Although ipilimumab only generated responses in approximately 10% of patients, it quickly became the standard of care. Additionally, it energized researchers to evaluate inhibiting other checkpoint pathways, as well as to investigate combinations of drugs that may increase response rates while maintaining long durations of response.3
Identifying new checkpoint pathway targets involves understanding the role of the immune system in cancer—more specifically, understanding the complex mechanisms that control tumor and immune interactions that can suppress tumor growth or stimulate it.6 T-cells are key components of adaptive immunity and have been described as soldiers who search out and destroy targeted invaders. On the basis of research completed in mouse models of cancer, T-cells appear to be the primary cells responsible for killing cancer cells and, consequently, are a target for immunotherapies against malignancies.3 Activating naïve T-cells to promote proliferation and differentiation to act against an infection or malignancy requires 2 signals (Figure 1)7: the first signal is antigen specific and occurs when a T-cell receptor binds with a major histocompatibility complex that is presenting an antigen peptide; this is followed by a second non-antigen specific costimulatory signal that causes CD28 receptors on T- cells to bind to a B7 molecule on antigen-presenting cells (APCs).3,6 However, once a T-cell is activated, it is not intended to be “on” forever. To help maintain immunologic homeostasis and limit autoimmune disease, T-cell activation is inhibited by checkpoint activation in the presence of chronic antigen exposure. The cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein-1 (PD-1) are different checkpoint pathways that appear important in tumor/immune interactions and, when activated, lead to exhausted T-cells.6 Checkpoint immune inhibitors act against CTLA-4 and PD-1 or PD-1 ligands (PD-L1) and, thus, inhibit the negative signals and reinvigorate exhausted T-cells.6
| Figure 1. Mechanisms of Action of Checkpoint Inhibitors (Anti-CTLA4 and Anti-PD-1)7 |
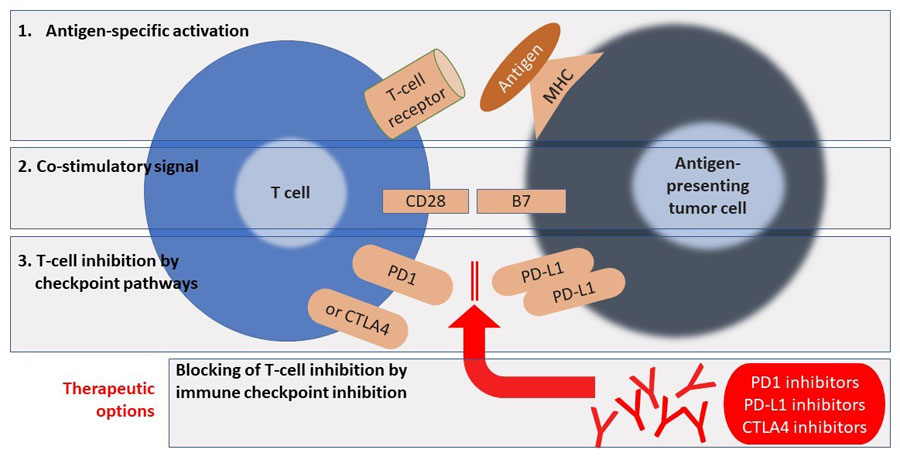 |
| Abbreviations: CTLA-4, cytotoxic T-lymphocyte-associated protein 4; MHC, major histocompatibility complex; PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand 1. |
Combining checkpoint inhibitors in clinical trials
In the past 3 years, 5 new checkpoint inhibitors have been approved by the FDA for use in cancers (Table 1).8-12 Approvals of these drugs were based on durable objective responses along with extended OS seen in patients with many different cancer types.13 Figure 2 shows the activities of PD-1 and PD-L1 inhibitors.14 Many trials are ongoing to determine the optimal uses for these agents, including the best combination therapies with other IO agents, chemotherapies, radiotherapies, and chemoradiotherapies.2 According to a recent analysis, 1105 trials investigating PD-1 or PD-L1 combination therapy have been started, and the vast majority include the 5 approved PD-1/PD-L1 agents; 49 of these trials are testing agents that have not yet been approved.2
| Table 1. Currently Approved Immune Checkpoint Inhibitors |
| Drug |
Dose |
Cancer indications |
Atezolizumab8
(PD-L1i) |
1200 mg IV over
60 min every 3
weeks |
Bladder, NSCLC |
Avelumab9
(PD-L1i) |
10 mg/kg IV over
60 min every 2
weeks |
Bladder, Merkel cell |
Durvalumab10
(PD-L1i) |
10 mg/kg IV over
60 min every 2
weeks |
Bladder, NSCLC |
Nivolumab11
(PD-1i) |
240 mg IV over 30
min every 2 weeks
or 480 mg IV over
30 min every 4
w
eeks* |
Bladder, colorectal (MSI-H/dMMR), head and neck squamous cell, hepatocellular, Hodgkin lymphoma, melanoma, NSCLC, renal cell |
Pembrolizumab12
(PD-1i) |
200 mg IV over 30
min every 3 weeks |
Bladder, gastric, head and neck, Hodgkin lymphoma, melanoma, MSI-H/dMMR, NSCLC |
*4-week regimen approved for melanoma, NSCLC, renal cell, Hodgkin lymphoma, head and neck squamous cell, bladder, and hepatocellular cancers. Starting doses are calculated as mg/kg for combination regimens with ipilimumab in melanoma and renal cell.
Abbreviations: IV, intravenously; MSI-H/dMMR, microsatellite instability-high/mismatch repair deficient; NSCLC, non-small cell lung cancer; PD-1i, programmed cell death protein-1 inhibitor; PD-L1i, programmed death-ligand 1 inhibitor. |
| Figure 2. PD-1/PD-L1 activity14 |
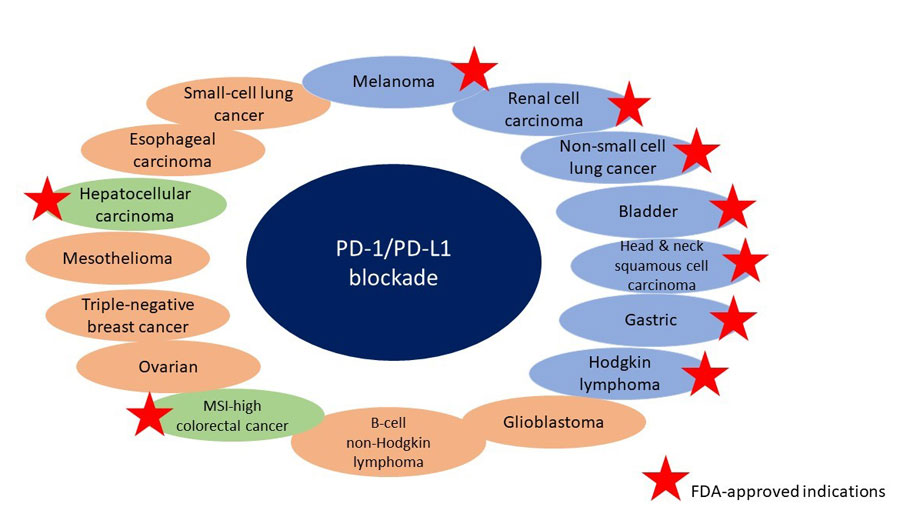 |
| Abbreviations: FDA, United States Food and Drug Administration; MSI, microsatellite instability; PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand 1. |
Combination therapies may improve responses through synergistic effects but still maintain the long duration of response seen with IO agents. Selecting combinations of drugs involves consideration of activity and mechanisms of resistance. For example, combining a precision medicine treatment, which can generate a very high, but short-lived, response with an IO therapy that generates a low, but long- lasting, response could bring out the best of both drug classes: a high response rate that is long lasting. Alternatively, combining 2 IO therapies that target different pathways could increase response rates and maintain the duration of response. Several combinations have been approved by the FDA for use in cancer. The IO and chemotherapy combination of pembrolizumab with pemetrexed and carboplatin is part of pembrolizumab’s indication for first-line use in NSCLC.14 The IO combination of nivolumab and ipilimumab has been approved by the FDA in unresectable or metastatic melanoma and advanced renal cell carcinoma.13 Many ongoing clinical trials are evaluating combinations of therapies that include an IO agent with the goal of improving median OS.14 In general, this approach to treatment could lead to cancer therapy in which 1 agent kills tumor cells, resulting in the release of tumor antigens and neoantigens, and an immune checkpoint inhibitor can then be used to “take the brakes” off of T-cells and provide long-term response.3
Bladder cancer
The development of anti-PD-1 and anti-PD-L1 therapies have changed the treatment landscape for bladder cancer. For 30 years, the most effective treatment for metastatic bladder cancer was combination chemotherapy, which was associated with a 5-year OS for all stages that ranged from 15% to 20%.15 All 5 of the checkpoint inhibitors have shown activity in bladder cancer, with response rates of 15% to 22% as second-line treatment in metastatic bladder cancer and 23% to 29% as first-line treatment in cisplatin-ineligible patients with metastatic bladder cancer. The efficacy is so obvious that single-arm trials led to the approval of 4 of the agents.15 Only 2 large randomized trials comparing chemotherapy to immunotherapy have been published to date.
In an international, open-label, phase 3 trial, 542 patients with advanced bladder cancer that had progressed after platinum-based therapy were randomized to the anti-PD-1 agent pembrolizumab or the investigator’s choice of chemotherapy with paclitaxel, docetaxel, or vinflunine. The median OS was significantly higher in the pembrolizumab group (n = 270) than in the chemotherapy group (n = 272) (10.3 months vs. 7.4 months, P = 0.002). In the pembrolizumab group, the most common treatment- related adverse events were pruritus (19.5%), fatigue (13.9%), and nausea (10.9%). The patients in the pembrolizumab group experienced fewer treatment-related adverse events of any grade than in the chemotherapy group.16
In a multicenter, open-label, phase 3, controlled trial, 931 patients with locally advanced or metastatic bladder cancer that had progressed with platinum-based chemotherapy were randomized to the anti-PD-L1 agent atezolizumab or the investigator’s choice of chemotherapy with vinflunine, paclitaxel, or docetaxel. Patients were stratified according to PD-L1 expression (< 1%, 1% to < 5%, and ≥ 5%). Interim results showed no significant difference in OS between the atezolizumab and chemotherapy groups (11.1 months vs. 10.6 months, P = 0.41)17; however, an exploratory analysis showed that high PD-L1 expression and high tumor gene expression were associated with improved outcomes in both chemotherapy and atezolizumab arms.18 The safety profile for atezolizumab was more favorable than for chemotherapy in terms of the rate of grade 3/4 treatment-related adverse events (20% vs. 43%) and adverse events leading to treatment discontinuation (7% vs. 18%).17
Melanoma
For patients diagnosed with early-stage melanoma, cure rates are very high: 5-year survival rates range between 92% and 97% for stage I and between 53% and 81% for stage II disease.19 However, for advanced disease—regional and distant metastatic melanoma—5-year survival ranges between 13% and 69% for stage III and is just 6% for stage IV disease. Identifying effective therapies for metastatic melanoma is of particular importance because of the lack of effective therapies for this disease and its rising incidence.7
Patient case 1: LB is a 45-year-old healthcare provider who has been diagnosed with metastatic melanoma (BRAF negative). She is the mother of 2 teenage daughters and hopes to have a chance of being cured, even if it requires participation in the most promising clinical trials.
Since 2011, 3 checkpoint inhibitors have been approved to treat metastatic melanoma. Ipilimumab is a CTLA-4 fully human monoclonal antibody that shows a 3-year OS rate of 26% in patients with treatment- naïve advanced melanoma and a 10-year OS rate of approximately 20%.20 When the anti-PD-1 agent nivolumab is combined with ipilimumab, the objective response rate (ORR) increases. In a double-blind phase 3 trial, 945 patients were randomized in a 1:1:1 ratio to receive nivolumab with ipilimumab, nivolumab alone, or ipilimumab alone. After 3 years of follow up, the median OS had not been reached in the nivolumab plus ipilimumab group; median OS was 37.6 months in the nivolumab alone group and 19.9 months in the ipilimumab alone group. The hazard ratio (HR) for death was 0.55 for nivolumab plus ipilimumab compared to ipilimumab alone (P < 0.001) and was 0.65 for nivolumab alone compared to ipilimumab alone (P < 0.001).20 Grade 3 or 4 treatment-related adverse events were seen in 59% of patients in the combination group, 21% in the nivolumab group, and 28% in the ipilimumab group. It is important to note that 39% of the combination group, 12% of the nivolumab group, and 16% of the ipilimumab group discontinued treatment due to toxicity.20 Although this trial demonstrated benefit with combined checkpoint inhibitors, some clinicians question whether the benefit is worth the increased toxicity. As seen in this trial, nivolumab is more effective than ipilimumab but has less toxicity. This is consistent with multiple trials evaluating a PD-1 inhibitor with a different checkpoint pathway inhibitor.
Indoleamine-2,3-dioxygenase-1 (IDO1) is a targetable enzyme in a different checkpoint pathway that has garnered significant research interest. Epacadostat is a potent and selective IDO1 inhibitor. Shown to play a role in fetal protection from the maternal immune system, IDO1 is overexpressed in many tumor types, including melanoma, ovarian, and colon cancers.21 Preclinical trials have shown that epacadostat has synergistic effects in combination with anti-PD-1 and anti-PD-L1 agents. Specifically, a combination of pembrolizumab and epacadostat in metastatic melanoma was promising: an interim report from a phase 3 trial of epacadostat plus pembrolizumab compared to pembrolizumab plus placebo stated the ORR in the epacadostat combination arm was 55% (29/53) with a progression-free survival (PFS) of 22.8 months. The most common adverse events of all grades for all patients were rash, fatigue, pruritus, arthralgia, diarrhea, nausea, and increased levels of aspartate transaminase, alanine transaminase, and lipase. However, only 6% of participating patients discontinued the treatment due to toxicity, which is a much lower rate than the almost 40% discontinuation rate for patients taking nivolumab plus pembrolizumab in the trial described above.21,22 Unfortunately, a recent press release announced that this trial failed to show benefit of epacadostat and the trial was closed to accrual.23 This combination continues to be studied in other tumors, but the excitement for this combination seems to be blunted. Indoximod is another IDO1 inhibitor that is being tested with pembrolizumab or nivolumab against melanoma, but the results of these investigations have yet to be reported.
Patient case 1: Potential treatments for LB include: nivolumab and ipilimumab, high-dose aldesleukin (interleukin [IL]-2), indoximod plus pembrolizumab or nivolumab, and stereotactic radiotherapy (15-20 Gy) plus nivolumab and ipilimumab. The nivolumab plus ipilimumab trial data is mature enough to conclude that ipilimumab is inferior to nivolumab with or without ipilimumab. The challenge with the combination is that 39% of patients discontinued treatment due to toxicity. The IDO1 inhibitor indoximod plus a PD-1 inhibitor appear to have synergy similar to that of nivolumab plus ipilimumab, yet the former is less toxic. Radiation with a CTLA-4 or PD-1 inhibitor appears to have synergy and animal data testing both IO therapies with radiation have shown significant activity. Many clinical trials testing radiation and IO therapy combinations are recruiting patients. The ongoing challenge is increased toxicity with additional therapies.
Non-small cell lung cancer
Most patients with non-small cell lung cancer (NSCLC) are diagnosed with regional or distant metastasis, and the 5-year survival rate is only 27% for those with regional disease and 4% for those with distant metastases.24 Owing to beneficial trial results, immune checkpoint inhibitors have become new standards of care for patients with advanced NSCLC. Current treatment recommendations include checkpoint inhibitors for specific patients with locally advanced NSCLC; checkpoint inhibitors can also be used as first-line or second-line treatment in metastatic NSCLC.25
Patient case 2: PJ is a 60-year-old man with newly diagnosed stage IV adenocarcinoma of the lung. His performance status is 1 and he is in relatively good health. Labs are normal or near normal. Pathology reports show no actionable mutation and low PD-L1 (tumor proportion score [TPS] of 8%). Which of the following regimens has the most efficacy according to clinical trial results?
- Cisplatin and pemetrexed
- Carboplatin, paclitaxel, and bevacizumab
- Carboplatin, paclitaxel, bevacizumab, and atezolizumab
The anti-PD-1 antibody nivolumab has been approved for second-line treatment of metastatic NSCLC that has progressed on or after platinum-based chemotherapy. Two international, open-label, phase 3 trials showed superior OS with nivolumab compared with docetaxel in patients with non-squamous26 or advanced squamous27 NSCLC who had progressed on prior therapy. These results were durable according to 2 years of follow-up data; more patients treated with nivolumab were alive than those treated with docetaxel whether the disease was advanced non-squamous NSCLC (n = 272, 29% vs. 16%) or advanced squamous NSCLC (n = 582, 23% vs. 8%). The median OS with nivolumab was 12.2 months for patients with non-squamous tumors and 9.2 months for those with squamous tumors; median OS was 9.5 months and 6 months for patients with non-squamous and squamous tumors treated with docetaxel, respectively.28 In both of these studies, nivolumab was associated with fewer all-grade and high-grade adverse events than docetaxel. The most frequent treatment-related adverse events seen with nivolumab in patients with squamous NSCLC were fatigue (16%), nausea (12%), decreased appetite (10%), and asthenia (10%). In patients with advanced squamous NSCLC, the most frequent treatment- related adverse events that occurred in patients who received nivolumab were fatigue (16%), decreased appetite (11%), and asthenia (10%).26,27
The anti-PD-1 antibody pembrolizumab has been approved for first-line use in patients with metastatic NSCLC whose tumors have a high percentage of viable cells expressing PD-L1 (TPS of 50% or higher) and with no epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumor aberrations.25 A randomized phase 3 study compared pembrolizumab to physician’s choice of platinum- based chemotherapy in 305 randomized patients with previously untreated advanced NSCLC with PD-L1 expression of 50% or higher.29 The median PFS was significantly higher in the pembrolizumab group than in the platinum-based chemotherapy group (10.3 vs. 6.0 months, P < 0.001). At 6 months, the estimated OS was 80.2% for the pembrolizumab group and 72.4% for the chemotherapy group (P = 0.005). Adverse events of any grade were less frequent in the pembrolizumab group (73.4% vs. 90.0%), as were grade 3 or higher events (26.6% vs. 53.3%).29
A phase 3 trial randomized 1202 patients with non-squamous metastatic NSCLC to 3 different treatment arms: atezolizumab with carboplatin and paclitaxel; atezolizumab with carboplatin, paclitaxel, and bevacizumab; and, carboplatin with paclitaxel and bevacizumab. The median PFS was 8.3 months in the atezolizumab/carboplatin/paclitaxel/bevacizumab arm and 6.8 months in the carboplatin/paclitaxel/bevacizumab arm (HR = 0.62; P < 0.0001).30 Interestingly, preliminary results showed that the substitution of bevacizumab with atezolizumab in the combination of carboplatin and paclitaxel did not lead to a significant OS benefit. Complete results are expected to be presented at the 2018 American Society of Clinical Oncology (ASCO) meeting.31 Treatment-related serious adverse events were observed in 25% of patients in the atezolizumab/carboplatin/paclitaxel/bevacizumab arm and 19% in the carboplatin/paclitaxel/bevacizumab arm.30
Patient case 2: All of the options are potential regimens for PJ, but clinicians should note that his TPS is only 8%, which is considered PD-L1 negative. The responses to these therapies may be modest, but the median OS in one trial was approximately 8 months. The addition of a PD-1 or PD-L1 inhibitor to chemotherapy in early phases of treatment improves PFS and may improve OS. Clinicians must remember that these chemo-immunotherapy combinations are only proven in adenocarcinoma; it is unclear if the combination will work in squamous histology. (Pemetrexed is not effective in squamous histology and bevacizumab is not safe in squamous histology).
EMERGING STRATEGIES FOR THE USE OF T-CELL MANIPULATION IN CANCER TREATMENT
In addition to checkpoint inhibitor therapy, other therapies are emerging that also target T-cells to treat cancers. Two technologies that include genetic engineering of cells have been shown to increase immune response.
Virus therapy
Talimogene laherparepvec (T-VEC) is a genetically engineered herpes simplex virus type 1 of the JS-1 strain that is administered intralesionally. It has a bimodal mechanism of action. First, it replicates and is oncolytic in cancer cells (local effect), but stress signals prevent it from replicating in normal cells; the second mechanism includes the translation of the inserted granulocyte-macrophage colony stimulating factor (GM-CSF) gene, which activates dendritic cells (professional APCs) to take up antigens and present them to T-cells, which creates a systemic immune response.32
In the phase 3 Oncovex (GM-CSF) Pivotal Trial in Melanoma (OPTIM), 436 patients with unresected stage IIB to IV melanoma were randomized (2:1) to receive intralesional T-VEC or subcutaneous GM-CSF. Among the 295 patients who received T-VEC, 64% of injected lesions (n = 2116), 34% of non-visceral lesions (n = 981), and 15% of uninjected visceral lesions (n = 177) decreased in size by at least 50%. In the same patients, complete resolution was seen in 47% of injected lesions, 22% of uninjected non-visceral lesions, and 9% of visceral lesions. In this group, 48 patients (approximately 16%) achieved durable responses.33
Chimeric antigen receptor-T-cell therapy
Chimeric antigen receptor (CAR)-T-cell therapy is a form of adoptive cellular therapy that is considered a “living drug.” CAR-T-cells are created by genetically engineering receptors that are expressed on the surface of T-cells. The genetic modification dictates the extracellular binding partners. For currently approved drugs, the extracellular portion binds to CD19, which is expressed on malignant B-cell cancers. The intracellular portion of the T-cell receptor has also been modified so it contains the second signal and is, thus, able to activate and direct cytotoxic T-cells to attack and kill tumor cells.34
Although CAR-T-cell therapy is directed towards tumor cells, it is associated with a potentially fatal systemic inflammation: cytokine release syndrome (CRS). The release of cytokines as part of the immune response is part of the normal process seen with activated T-cells. When CAR-T-cells are stimulated— particularly in patients with more extensive disease—a massive release of cytokines into the patient’s bloodstream can lead to high fevers, flu-like symptoms, and even shock.34 The timing of development of CRS varies, with a median onset of 2 to 3 days. Patients with a high pretreatment tumor burden may be at higher risk of developing CRS, but all patients should be monitored for symptoms. CRS can be effectively treated with standard supportive care measures and the anti-IL-6 monoclonal antibody, tocilizumab. Corticosteroids are also effective, but their lymphotoxic properties generally dictate that they are reserved for tocilizumab-refractory toxicity or neurotoxicity due to cerebral inflammation.35
Tisagenlecleucel is the first FDA-approved CAR-T-cell therapy; it targets CD19, which is commonly found on B-cell malignancies. In a phase 1-2a study of tisagenlecleucel in 60 children and young adults with relapsed or refractory acute lymphoblastic leukemia (ALL), the rate of complete remission was 93%.36 In a phase 2 study of 75 patients with CD19-positive relapsed or refractory B-cell ALL, the overall remission rate was 81% within 3 months. All patients with a response to treatment were negative to minimal residual diseases. At 12 months, the rate of OS was 76% (95% confidence interval, 63% to 86%) and median duration of remission was not reached. Grade 3/4 adverse events that were believed to be treatment related occurred in 73% of patients, and 77% of patients developed CRS.36
PRACTICAL CHALLENGES WITH IO TREATMENTS
Despite the benefits seen with IO agents, they present challenges in clinical practice. Evaluating response and managing safety concerns are difficult and require special consideration by the healthcare team, as well as education for patients and caregivers.
Measuring response
The timing of tumor response to IO agents is different than what is seen with chemotherapy. As a result, the traditional systems of assessment have been revised for IO agents. The Response Evaluation Criteria in Solid Tumors (RECIST) is the standard tool for evaluating response in solid tumor trials and is used clinically to determine when cytotoxic treatment has failed.37 RECIST uses a unidimensional measure: the longest diameter of target tumor lesions is used to determine progressive disease, stable disease, partial response, and complete response. These criteria have been shown to be reproducible and remain the standard for evaluating response in clinical trials.38 Still, while RECIST continues to be used in IO clinical trials, the criteria are not clinically useful due to the altered pattern of response. Patients receiving IO therapies commonly experience a pseudoprogression, which occurs when tumor growth occurs after treatment initiation but before a treatment response. A pseudoprogression is consistent with the mechanism of action of IO agents, which require time to generate an effective immune response. As a result, the RECIST criteria will define some patients with disease progression who will eventually experience durable responses.37 For this reason, clinicians do not stop IO therapy after initial documentation of tumor growth (which would necessitate discontinuation according to RECIST criteria).39
To address these challenges, the immune-related response criteria (irRC) have been developed to recognize the response patterns observed with IO agents.39 The irRC uses bidimensional measurement and accounts for potential pseudoprogression.40 As shown in Table 2, the irRC criteria require confirmation of progressive disease through a second confirmatory scan at 4 weeks or later.40
| Table 2. Differences Between RECIST and irRC40 |
| |
RECIST
Unidimensional measurement |
irRC
Bidimensional measurement |
| Complete response (CR) |
Disappearance of all lesions |
| Partial response (PR) |
≥ 30% decrease in tumor burden compared with baseline* |
≥ 50% decrease in tumor burden compared with baseline* |
| Stable disease (SD) |
Not PR, CR, or PD |
| Progressive disease (PD) |
≥ 20% +5-mm absolute increase in tumor burden compared with nadir
Appearance of new lesions or progression of non-target lesions |
≥ 25% increase in tumor burden compared with baseline, nadir, or reset baseline*
New lesions added to tumor burden* |
* Confirmation required: next scan ≥ 4 weeks later.
Abbreviations: irRC, immune-related response criteria; RECIST, Response Evaluation Criteria in Solid Tumors, v 1.1. |
Predicting response to IO agents
Identifying biomarkers that are predictive of response to IO treatments are crucially important because immunotherapies may only benefit a small proportion of patients and can be associated with acute toxicities.41 Ideally, a biomarker will predict who will and who will not respond to treatment. Realistically, though, there are no perfect biomarkers, but, before withholding treatment, biomarkers can be strong predictors in identifying patients who will not benefit from treatment compared to standard therapy. On the basis of the pharmacology, using PD-L1 expression on tumors seems like an ideal biomarker, and such testing has been performed with all the PD-1 and PD-L1 inhibitors in clinical trials. Expression of PD-L1 has been prognostic, in general, but PD-L1-negative patients also respond to treatment. The challenges have been the variations in tumor tissue, assays used, and thresholds to determine positivity or negativity. Currently, PD-L1 expression is only a valuable biomarker to select first-line pembrolizumab in advanced stage NSCLC. The FDA approved the cobas assay that utilizes the 22c3 monoclonal antibody for immunohistochemistry with a TPS of 50% or greater for PD-L1, which is estimated to be found in approximately 30% of patients with advanced NSCLC. Selecting these PD-L1- positive patients demonstrated that pembrolizumab monotherapy was more effective than chemotherapy for first-line treatment of advanced or metastatic adenocarcinoma NSCLC.33 However, PD-L1 expression is not predictive in all cancers that benefit from treatment with PD-1 inhibitors. A study in advanced renal cell carcinoma showed that PD-L1 expression of 1% or greater was found in roughly one-quarter (24%) of patients. These patients were randomized to nivolumab or everolimus; PD- L1 expression was not shown to correlate with response. Despite this finding, nivolumab was associated with longer OS and fewer grade 3/4 adverse events than the comparator arm.42
Other factors may be used to predict response to immunotherapy. The total number of mutations found in a tumor specimen—or the mutation load or tumor mutational burden—can be a predictor of response to immunotherapy. This may be because tumors with a high mutational load may harbor more neoantigens, which may make the tumors recognizable to the immune system and make them targets for checkpoint inhibitor therapy.43 Human cancers are known to have high rates of mutation and those rates can increase when cellular pathways such as DNA damage repair and replication are involved, as well as after exposure to environmental factors such as tobacco smoke and ultraviolet light.43
Mutational burden ranges widely in patients with NSCLC. Those with a history of smoking generally have more somatic mutations than those who never smoked. A study of 2 independent cohorts of patients with NSCLC treated with pembrolizumab showed that the mutational burden correlated with objective clinical response, durable clinical benefit, and PFS. The evaluation determined that durable clinical benefit was achieved in 75% of patients with a nonsynonymous mutational burden of 178 or higher and only 14% of those with a mutational burden less than 178.44 Another study evaluated the mutational burden in 64 patients with malignant melanoma who had been treated with CTLA-4 blockade. There was a significant correlation with mutational load and degree of clinical benefit, but that alone was not predictive of benefit. The study then identified a “neoantigen landscape” in these patient cohorts that was associated with a strong response to treatment with CTLA-4 blockade.45 This area of research will continue to be of importance in the development of IO agents.
Unique safety profiles of IO agents
The safety profiles of IO agents differ from those for chemotherapy agents and other therapies because of their immune-related mechanism of actions. In particular, immune checkpoint inhibitors can cause T- cell reactivation/proliferation, which can produce immune-related adverse events (irAEs) that resemble autoimmune conditions. The recognition of the special considerations required for IO agents led to the development of a set of guidelines by ASCO in collaboration with the National Comprehensive Cancer Network.46 These guidelines were developed after a systematic review of available literature, but the authors noted that there is a lack of high-quality evidence in the management of irAEs. As a result, the treatment recommendations are based on expert consensus.46 Similar guidelines have also been developed by the European Society for Medical Oncology.47
While irAEs can manifest in any organ system, they are most commonly seen in the skin, gastrointestinal tract, and lungs, as well as the endocrine, thyroid, adrenal, pituitary, musculoskeletal, renal, nervous, hematologic, cardiovascular, and ocular systems.45 The majority of patients will likely experience some type of side effect, but only about 10% of these events will be grade 3 or 4.48 The timing of the development of these side effects varies, but they usually appear 8 to 12 weeks after the start of treatment.47,48
The ASCO guidelines provide specific recommendations for each organ system but have also provided general recommendations about the management of irAEs (Table 3).46 In general, immune checkpoint inhibitor therapy can be continued with symptom management and close monitoring for grade 1 events. For grade 2 or 3 toxicity, immune checkpoint inhibitors should be temporarily discontinued and steroids should be initiated; the checkpoint inhibitor can be restarted when symptoms revert to grade 1 or lower and steroids can be tapered to a daily prednisone dose of 10 mg or less. For grade 4 toxicities, immune checkpoint therapy should be permanently discontinued. The only exception is endocrinopathies, which can be treated and the checkpoint therapy can be continued. The recommendations note that when new symptoms appear, they should be suspected to be treatment related.46
| Table 3. General Recommendations for the Treatment of Immune-related Adverse Events*46 |
| Grade 1 |
Continue immune checkpoint inhibitor therapy
Monitor for symptoms every 2 to 3 days |
| Grade 2 |
Suspend immune checkpoint inhibitor therapy
Consider restarting therapy when symptoms return to grade 1
Corticosteroids may be started: prednisone or methylprednisolone 0.5 mg/kg/day |
| Grade 3 |
Suspend immune checkpoint inhibitor therapy
High-dose corticosteroids may be started: prednisone or methylprednisolone 1-2 mg/kg/day
Taper over 4-6 weeks
Additional immune suppression for refractory patients |
| Grade 4 |
Discontinue immune checkpoint inhibitor therapy and hospitalize patient
High-dose corticosteroids may be started: prednisone or methylprednisolone 1-2 mg/kg/day
Taper over 4-6 weeks
Additional immune suppression for refractory patients |
| *Clinicians should always consider organ-specific recommendations in addition to those provided in the guidelines. |
Knowledge of potential irAEs is critical to ensure patient safety and optimize outcomes, so it is important that all members of the healthcare team, as well as patients and caregivers, are educated about the potential adverse events seen with immunotherapy.45,46 In fact, the ASCO guidelines recommend that “patients and family caregivers should receive timely and up-to-date education about immunotherapies, their mechanism of action, and the clinical profile of possible irAEs before initiating therapy and throughout treatment and survivorship.”46 Pharmacists are uniquely positioned to provide this important education.
CONCLUSION
New drugs are continually being investigated to improve outcomes in cancer. Pharmacists play a key role in integrating these new drugs safely and effectively into routine practice. With unique mechanisms of action, IO agents and the resulting combination regimens have novel side effect profiles that require education about appropriate monitoring and therapeutic management for all members of the healthcare team. As more of these IO agents are introduced, the role of pharmacists in balancing safety and efficacy through education, patient monitoring, and diagnosis and management of irAEs will continue to grow.
November 21, 2018 Quarterly Review
Since the publication of the monograph, the following has occurred:
- Nivolumab has been approved for metastatic small cell carcinoma of the lung
- Pembrolizumab has been approved for hepatocellular carcinoma
- The IO combination, pembrolizumab and carboplatin and either paclitaxel or nab-paclitaxel has been approved for metastatic squamous NSCLC
- Cemiplimab-rwlc has been approved for metastatic or locally advanced squamous cell carcinoma of the skin
- Tisagenlecleucel has been approved for relapsed or refractory diffuse large B-cell lymphoma
|
August 8, 2018 Quarterly Review
Since the publication of the monograph the following has occurred:
- Pembrolizumab has been approved for cervical cancer and primary mediastinal large B-cell lymphoma
- The IO combination, nivolumab and impilimumab has been approved for metastatic colorectal cancer with MSI-H/dMMR
- Complete trial results for the phase 3 trial—non-squamous metastatic NSCLC to 3 different treatment arms: atezolizumab with carboplatin and paclitaxel; atezolizumab with carboplatin, paclitaxel, and bevacizumab; and, carboplatin with paclitaxel and bevacizumab—were reported at ASCO in June. The results indicated that the addition of atezolizumab to bevacizumab plus chemotherapy significantly improved progression-free survival and overall survival among patients with metastatic nonsquamous NSCLC, regardless of PD-L1 expression and EGFR or ALK genetic alteration status.1
1Socinski M, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. NEJM; 2018; 378: 2288-2301.
|
February 12, 2019
Since the publication of the monograph the following has occurred:
- Pembrolizumab has been approved for the treatment of recurrent locally advanced or metastatic Merkel cell carcinoma.
- Atezolizumab in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of patients with metastatic non-squamous, non-small cell lung cancer with no EGFR or ALK genomic tumor aberrations has been approved.
|
REFERENCES
- Richardson MA, Ramirez T, Russell NC, Moye LA. Coley toxins immunotherapy: a retrospective review. Altern Ther Health Med. 1999;5(3):42-7.
- Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol. 2018;29(1):84-91.
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205-14.
- Hashimoto M, Kamphorst AO, Im SJ, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. 2018;69:301-18.
- Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889-94.
- Daud A. Current and emerging perspectives on immunotherapy for melanoma. Semin Oncol. 2015;42 Suppl 3:S3-S11.
- Karlsson AK, Saleh SN. Checkpoint inhibitors for malignant melanoma: a systematic review and meta-analysis. Clin Cosmet Investig Dermatol. 2017;10:325-39.
- Atezolizumab [package insert]. South San Francisco, CA: Genentech, Inc.;2018.
- Avelumab [package insert]. Rockland, MD: EMD Serono, Inc.;2017.
- Durvalumab [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP;2018.
- Nivolumab [package insert]. Princeton, NJ: Bristol-Myers Squibb Company;2018.
- Pembrolizumab [package insert]. Whitehouse Station, NJ: Merck & Co, Inc;2017.
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275-87.
- Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139-48.
- Aggen DH, Drake CG. Biomarkers for immunotherapy in bladder cancer: a moving target. J Immunother Cancer. 2017;5(1):94.
- Bellmunt J, de Wit R, Vaughn DJ, et al; KEYNOTE-045 Investigators. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015-26.
- Powles T, Duran I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2018;391(10122):748-57.
- Powles T, Loriot Y, Ravaud A, et al. Atezolizumab (atezo) vs. chemotherapy (chemo) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC): Immune biomarkers, tumor mutational burden (TMB), and clinical outcomes from the phase III IMvigor211 study. J Clin Oncol. 2018;36(6 suppl);409.
- American Cancer Society. Survival rates for melanoma skin cancer, by stage. https://www.cancer.org/cancer/melanoma-skin-cancer/detection-diagnosis-staging/survival-rates-for-melanoma-skin-cancer-by-stage.html. Updated May 20, 2016. Accessed May 20, 2018.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345-56.
- Yue EW, Sparks R, Polam P, et al. INCB24360 (epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett. 2017;8(5):486-91.
- Hamid O, Gajewski T, Frankel A, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma: phase 1 and 2 efficacy and safety results from ECHO-202/KEYNOTE-037. Presented at 2017 ESMO Congress; September 8-12, 2017; Madrid, Spain.
- Incyte and Merck Provide Update on Phase 3 Study of Epacadostat in Combination with KEYTRUDA® (pembrolizumab) in Patients with Unresectable or Metastatic Melanoma [press release]. Wilmington, DE and Kenilworth, NJ: Incyte Corporation and Merck Corporation; April 6, 2018. https://www.businesswire.com/news/home/20180406005141/en/Incyte-Merck-Provide-Update-Phase-3-Study. Accessed May 22, 2018.
- American Cancer Society. Cancer Facts & Figures 2018. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf. Published 2018. Accessed May 22, 2018.
- National Comprehensive Cancer Network. Non-small cell lung cancer. Version 4.2018. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Published April 26, 2018. Accessed May 22, 2018.
- Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627-39.
- Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123-35.
- Horn L, Spigel DR, Vokes EE, et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017;35(35):3924-33.
- Reck M, Rodriguez-Abreu D, Robinson AG, et al; KEYNOTE-024 Investigators. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823-33.
- Reck M, Socinski MA, Cappuzzo F, et al. Primary PFS and safety analyses of a randomized phase III study of carboplatin + paclitaxel +/− bevacizumab, with or without atezolizumab in 1L non-squamous metastatic NSCLC (IMpower150). Ann Oncol. 2017;28(11):Abstract LBA1_PR.
- The ASCO Post. IMpower150: Increased OS with atezolizumab/bevacizumab plus chemotherapy in advanced nonsquamous NSCLC. http://www.ascopost.com/News/58689. Published April 3, 2018. Accessed April 22, 2018.
- Ott PA, Hodi FS. Talimogene laherparepvec for the treatment of advanced melanoma. Clin Cancer Res. 2016;22(13):3127-31.
- Andtbacka RH, Ross M, Puzanov I, et al. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann Surg Oncol. 2016;23(13):4169-77.
- National Cancer Institute. CAR T Cells: Engineering patients' immune cells to treat their cancers. https://www.cancer.gov/about-cancer/treatment/research/car-t-cells. Updated December 14, 2017. Accessed April 28, 2018.
- Shank BR, Do B, Sevin A, et al. Chimeric antigen receptor T cells in hematologic malignancies. Pharmacotherapy. 2017;37(3):334-45.
- Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-48.
- Seymour L, Bogaerts J, Perrone A, et al; RECIST working group. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18(3):e143-52.
- Nishino M, Giobbie-Hurder A, Gargano M, et al. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin Cancer Res. 2013;19(14):3936-43.
- Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412-20.
- Hodi FS, Hwu WJ, Kefford R, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J Clin Oncol. 2016;34(13):1510-7.
- Desrichard A, Snyder A, Chan TA. Cancer neoantigens and applications for immunotherapy. Clin Cancer Res. 2016;22(4):807-12.
- Motzer RJ, Escudier B, McDermott DF, et al; CheckMate 025 Investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803-13.
- Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34.
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124-8.
- Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189-99.
- Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018:JCO2017776385.
- Haanen JBAG, Carbonnel F, Robert C, et al; ESMO Guidelines Committee. Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2017;28(suppl 4): iv119-42.
- Champiat S, Lambotte O, Barreau E, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2016;27(4):559-74.
Back to Top