
Expired activity
Please go to the PowerPak
homepage and select a course.
Biosimilars in Oncology: Current Status, Future Directions, and Clinical Considerations
INTRODUCTION
Biologic therapies – agents derived from living sources - have revolutionized the treatment of oncology patients, leading to substantial improvements in outcomes in a variety of disease states.1 However, due to the unique complexity of manufacturing and development of these therapies, the cost of biological products is substantial. For example, the wholesale acquisition cost (WAC) for inotuzumab ozogamicin, a therapy approved by the United States (US) Food and Drug Administration (FDA) for treatment of acute lymphoblastic leukemia, is approximately $75,000 for one cycle in a patient with 2 m2 of body surface area (BSA).2
While competition from generic drug manufacturers helps to curtail prices of higher-cost small molecule medications, it is not possible to manufacture “generic” biologic therapies that are molecularly identical to the originator product. Instead, biosimilar products have become the avenue for reducing the high cost of such therapies, improving patient access and offering additional options for clinicians. A biosimilar is a biologic product that is highly similar to and has no clinically meaningful differences from the FDA-approved reference product in terms of safety, purity, and potency (safety and effectiveness).3
This review highlights the requirements, key concepts, and challenges associated with biosimilar products in oncology, including a discussion of approved biosimilar products and those in later stages of development.
Biologic vs. Small Molecule Therapies
Small molecule therapies (e.g., aspirin, imatinib) are produced via standard chemical synthesis and have reproducible, consistent structures that are generally smaller than 1000 daltons. This enables generic manufacturers to reproduce identical molecules via the same chemical synthesis strategy. On the other hand, biologic therapies are produced by living systems – typically using recombinant DNA technology. The DNA sequence of a therapeutic protein is introduced into a living host cell (e.g., bacteria, yeast, or mammalian cells), allowing for the transcription and product of the protein of interest on a larger scale. These therapeutic proteins are large structures with masses up to hundreds of thousands of daltons and complex secondary, tertiary, and quaternary structures. Accordingly, biologic molecules are sensitive to slight changes to the manufacturing and handling processes, which can lead to variations, even from batch to batch, in post-translational protein modifications (e.g., acetylation, phosphorylation, glycosylation), and the tertiary and quaternary structure of the protein. The result of these changes is wide ranging – while many minor modifications are inconsequential, some modifications may lead to reduced effector activity of the biologic therapy, a change in pharmacokinetics of the product, or an increase in immunogenicity of the protein. Because many of these therapeutic proteins are of nonhuman origin/structure, immunogenicity is of particular concern. The production of antibodies against the therapeutic protein may lead to accelerated clearance of the product, a lack of efficacy, or in some cases, severe allergy or anaphylaxis.3–5
Approval Pathways for Biosimilars
To demonstrate generic equivalence, small molecule therapies have to demonstrate that they contain the same active ingredient as and demonstrate bioequivalence to the reference product through a pharmacokinetic study.3–5 Given the aforementioned complexities of biosimilar manufacturing, it is not possible for biosimilars to have a perfectly identical structure to a reference biologic product. Therefore, it is not possible to create a generic version of the protein in the same sense as with a small molecule drug. Furthermore, no single study can establish biosimilarity.
The Biologics Price Competition and Innovation (BPCI) Act of 2009 was passed as a part of the Patient Protection and Affordable Care Act to provide an appropriate approval pathway for biosimilar medications.4 Under the BPCI Act, the FDA has established a stepwise, “totality of the evidence” approach in which multiple analyses are necessary to approve a biosimilar medicine.4 This approach starts with extensive physicochemical and functional analyses, followed by pharmacokinetic and pharmacodynamics (PK/PD) studies, evaluation of immunogenicity, and limited clinical trials with head-to-head comparisons and sensitive endpoints (e.g., biomarkers and measures that can show safety and/or effectiveness) in a sensitive patient population (e.g., for a head-to-head trial, treatment-naive patients).4
Unlike the full biological reference product approval process, which focuses on demonstrating safety and efficacy, the biosimilar approval process focuses on demonstrating the highly similar nature of a biosimilar agent to the reference product. The intent is not to repeat studies of safety and efficacy, but to demonstrate that the biosimilar will produce the same clinical results as the reference product in an abbreviated pathway. Because of this, the focus shifts to a heavy emphasis on analytical and preclinical characterization as the foundation for biosimilar approval (Figure 1). At each step of the development process, FDA works with the manufacturer of the biosimilar to evaluate similarity and to plan the next evaluation to remove residual uncertainty about whether the biosimilar will provide similar clinical efficacy and safety in patients.
Figure 1. Foundation of Biosimilarity Data
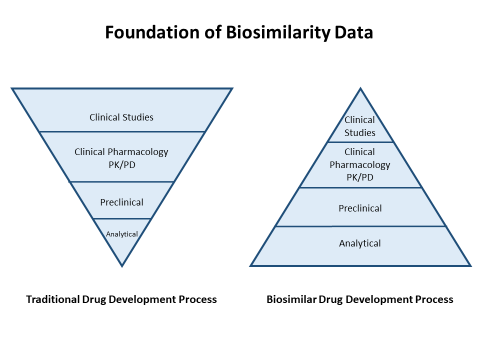 |
Compared with Europe and other countries, a unique aspect of the FDA approval process for biosimilars is the creation of a separate category of approval termed an “interchangeable biosimilar.” Interchangeability is intended to permit the substitution of a biosimilar product for the reference product without the intervention of the prescriber, similar to generic substitution with small molecule generic drugs. An interchangeable product “can be expected to produce the same clinical result as the reference product in any given patient”; the sponsor must demonstrate that “the risk to efficacy and safety with alternating or switching between the two products is not greater versus consistent use of the reference product.”5 Because the draft guidance on interchangeability was not issued by FDA until January 2017, biosimilar manufacturers were not able to pursue the interchangeability designation prior to that time. Since no biosimilars have been approved by FDA since that time, none of the products currently approved in the US are interchangeable. This does not imply that they are inferior to products marketed later that have the interchangeable biosimilar designation, but rather that the designation did not exist when they were approved, and they have not undergone the studies necessary to demonstrate that substitution can occur without the involvement of the prescriber.
Even though none of the current US- or European-approved biosimilars has an “interchangeable” designation, the safety of single switches between the reference product and a biosimilar is supported by a growing body of evidence.6 In the US, state regulations generally require that pharmacists notify the prescriber when substitution of a biosimilar occurs. Thus, pharmacists must contact prescribers to change a prescription from the reference product to a new prescription for the noninterchangeable biosimilar.7
Another key component of the biosimilar abbreviated approval pathway is the concept of indication extrapolation. Extrapolation has been one of the most contentious issues with the introduction of biosimilars. It refers to the regulatory process that permits the extrapolation of data from one indication to support licensing for other indications that have not been directly studied in clinical trials.4 Extrapolation reduces the need to conduct clinical studies across multiple patient populations and conditions; the creation of such an abbreviated pathway is based on the assumption this approach is rational and viable given what is known about the mechanism of action, site of action, and potential differences between patient populations in response or immunogenicity profiles. For example, for the approval of biosimilar infliximab, studies were conducted in patients with rheumatoid arthritis and ankylosing spondylitis comparing the biosimilar with the reference infliximab.8,9 Based on the results of these studies, regulators in both the US and Europe granted approval covering other indications that were not specifically studied.8–10 To date, all biosimilars that have been approved by the European Medicines Agency (EMA) and FDA have been approved for all of the eligible indications of the reference products. Within oncology, products are often commonly used for indications and in a manner that are not specifically mentioned in the license. Therefore, formulary committees will need to consider the issue of extrapolation of the use of a biosimilar in practice to scenarios where the reference product may be used for nonapproved indications.
The naming convention for biosimilars has also been a source of contention. Some have advocated that biosimilars should share the same international nonproprietary name (INN) as the reference product. This would facilitate the adoption of biosimilars in practice and would be consistent with how small molecule generic products are named. Others have argued that biosimilars are not “generic” in the sense that they cannot be completely identical and that they have greater potential for immunogenicity than small molecule drugs. For this reason, they need to have distinct names to help facilitate pharmacovigilance and to prevent inappropriate product switching. FDA has chosen a naming convention in which the biosimilars will share the same base name as the reference product, but will be distinguished by the addition of a four-character suffix that is “devoid of meaning.”11 Existing and new biological reference products will be renamed with a suffix as well. The intent is that the suffix will facilitate differentiation and pharmacovigilance efforts. This decision has been criticized in that it could inhibit the adoption of biosimilars by implying that there are important differences between the biosimilar and the reference product that require the need for different names, and also that the “nonmemorable” nature of the four-character suffix could actually make it more difficult for patients and providers to clearly communicate about which product is intended. Some have advocated for the use of brand names in addition to the INN-suffix to assure clarity in communication about biosimilars and reference biological products.12
Current Biosimilar Landscape
FDA had approved 12 biosimilar products at the time this program was prepared in November 2018; many more were under development and consideration.3 Of these, 7 biosimilar products provide potential alternatives for 5 reference biological products with oncology-related indications (Table 1).3,13–24 The majority of these – filgrastim-sndz (Zarxio), filgrastim-aafi (Nivestym), pegfilgrastim-jmdb (Fulphila), pegfilgrastim-cbqv (Udenyca), and epoetin alfa-epbx (Retacrit) – are supportive care medications, while bevacizumab-awwb (Mvasi) and trastuzumab-dkst (Ogivri) are approved for the treatment of oncologic conditions. Not all of the approved biosimilar products are marketed in the United States because of patent litigation and other factors.1
| Table 1. FDA-Approved Biosimilar Products With Oncology-Related Indications |
| Biosimilar (trade name) Pharmaceutical Co. |
FDA Approval Date |
Indications |
Notes |
Filgrastim-sndz (Zarxio)
Sandoz |
March 2015 |
- Patients receiving myelosuppressive chemotherapy
- Patients receiving induction and/or consolidation chemotherapy for acute myelogenous leukemia
- Patients undergoing bone marrow transplant
- Mobilization for autologous hematopoietic progenitor cell collection
- Patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia
- Patients acutely exposed to myelosuppressive doses of radiation
|
- Cannot accurately deliver doses to patients with body mass <36 kg because only prefilled syringes are available
|
Filgrastim-aafi (Nivestym)
Hospira/Pfizer |
July 2018 |
- Patients receiving myelosuppressive chemotherapy
- Patients receiving induction and/or consolidation chemotherapy for acute myelogenous leukemia
- Patients undergoing bone marrow transplant
- Mobilization for autologous hematopoietic progenitor cell collection
- Patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia
|
- Available in both prefilled syringes and vials
|
Pegfilgrastim-jmdb (Fulphila)
Mylan |
June 2018 |
- Patients receiving myelosuppressive chemotherapy
|
- Available only in prefilled syringes; not designed for direct administration of doses less than 0.6 mL (6 mg)
- No on-body injector available
|
Pegfilgrastim-cbqv (Udenyca)
Coherus Biosciences |
November 2018 |
- Patients receiving myelosuppressive chemotherapy
|
- Available only in prefilled syringes; not designed for direct administration of doses less than 0.6 mL (6 mg)
- No on-body injector available
|
Epoetin alfa-epbx (Retacrit)
Hospira/Pfizer |
May 2018 |
Same as reference product
- Treatment of anemia due to
- Chronic kidney diseases
- Zidovudine in patients with human immunodeficiency virus infections
- Concomitant myelosuppressive chemotherapy
- Reduction of allogeneic red blood cell transfusions in patients undergoing elective, noncardiac, nonvascular surgery
|
|
Bevacizumab-awwb (Mvasi)
Amgen |
Sep 2017 |
- Metastatic colorectal cancer
- First line nonsquamous, non-small-cell lung cancer
- Progressive glioblastoma
- Metastatic renal cell carcinoma
- Persistent, recurrent, or metastatic cervical cancer
|
- Not approved for epithelial ovarian, fallopian tube, or primary peritoneal cancer due to orphan exclusivity protection of US-reference bevacizumab
|
Trastuzumab-dkst (Ogivri)
Mylan |
Dec 2017 |
Same as reference product
- Adjuvant breast cancer
- Metastatic breast cancer
- Metastatic gastric cancer
|
|
| Sources: Compiled from references 3 and 13–24. |
In contrast with the situation in the US, the use and adoption of biosimilars in Europe has been more extensive. More than 40 biosimilars have been marketed in Europe — including some for 12 years or more — these have more than 20 approved oncologic indications. Reassuringly, no biosimilar products approved by the EMA have subsequently been removed from the market due to concerns with efficacy or safety.1
One of the major advantages of biosimilars in oncology is reducing the high financial burden that biologic therapies place on patients and healthcare systems. The cost of generic small molecule medications is generally significantly lower than branded products (typically 70% to 80% lower). Biosimilars do offer a price reduction, although it is not as large as with generics (typically a 30% price reduction).25 The economic impact of biosimilars is challenging to estimate, as it depends on the uptake and market share of biosimilars in clinical practice, the reimbursement rate for biosimilars, and other pricing considerations.26,27
As an example of the positive impact of biosimilars, in Europe, biosimilar epoetins have an estimated market share of more than 25% across 10 countries. In Germany, the availability of biosimilar erythropoiesis-stimulating agents (ESAs) provided a saving of 60 million euros during their first year on the market.28 Data in the United States on the full economic impact of biosimilars is still maturing but is promising. A pharmacoeconomic model estimated a 5-year savings of $256 million for the use of biosimilar filgrastim in patients receiving myelosuppressive chemotherapy.26 A RAND analysis estimated the use of biosimilars in the US will lead to a reduction in direct spending on biologic drugs of $54 billion from 2017 to 2026, which is approximately 3% of the total estimated biologic spending over this time period.29
To continue to incentivize the use of biosimilars in the outpatient oncology space, the Centers for Medicare and Medicaid Services has decided that approved biosimilars with a common reference product will be coded separately and reimbursed at the average sales price (ASP) of the biosimilar product plus 6% of the reference product ASP, providing a small incentive to the use of lower cost biosimilars in this setting.1 Given the rapid rise in biologic costs in oncology over the last decade, the use and investigation of lower cost biosimilar medications is urgently needed.
Case Study 1
As an oncology pharmacist in an integrated health system, you are involved in formulary decisions and product interchange policies. The drug spend for the filgrastim reference product in your institution is currently $7 million per year. The pharmacy and therapeutics committee is considering a policy of preferred use of a biosimilar filgrastim product. One physician has expressed opposition, stating that “the biosimilars just aren’t the same” as the innovator product and that the one being considered is not approved by FDA as “interchangeable.” What are two or three possible lines of reasoning that you can use to help convince this practitioner that biosimilars should be considered?
Among the possible approaches that could be used to ground the discussion in facts are the stepwise biosimilar approval pathway used at FDA, the later development of a pathway for biosimilars to be recognized as interchangeable, published literature documenting years of clinical experience with biosimilars in Europe, and the substantial cost reductions to patients, payers, and the institution with use of biosimilars – as much as $2.1 million annually for this one agent based on published figures and the amount currently spent on originator brand of filgrastim. |
FDA-Approved Biosimilars in Oncology
Biosimilar filgrastim products
Filgrastim (Neupogen), the reference biologic, is a white blood cell growth factor (granulocyte colony-stimulating factor [G-CSF]) that is currently approved for cancer patients receiving myelosuppressive chemotherapy or myelosuppressive radiation, for patients with acute myeloid leukemia receiving induction or consolidation chemotherapy, for cancer patients receiving a bone marrow transplant, for patients undergoing peripheral blood progenitor cell collection and therapy, for patients with severe, chronic neutropenia. Filgrastim is typically given daily at 5–10 mcg/kg/day (depending on indication) to stimulate granulocyte production.22
Filgrastim-sndz (Zarxio) was approved by the FDA in March 2015. Manufactured by Sandoz, Inc., Zarxio was the first biosimilar approved by FDA. Filgrastim-sndz was approved for the all but one of the indications as the reference biologic (for patients acutely exposed to myelosuppressive doses of radiation), based on the totality of the evidence. The radiation exposure indication was granted to the reference biologic under orphan drug designation in 2015 as a medical countermeasure in the case of a radiological/nuclear incident, based on animal data according to the Animal Rule.30 Thus, this indication is not patent-eligible for the biosimilars to seek approval. FDA approval took into consideration the extensive preclinical work to demonstrate an identical amino acid sequence, highly similar higher-order structure, and highly similar batch-to-batch product variations to the reference biologic.31 Filgrastim-sndz was also shown to be bioequivalent to the reference biologic, as well as to have a similar PD profile through demonstration of similar absolute neutrophil count (ANC) and CD34+ cell counts (a marker for mobilization indications) in a randomized, double-blind cross-over study in healthy volunteers (Study EP06-109).23 One of the pivotal clinical studies that led to FDA approval of filgrastim-sndz was Study EP06-302, a randomized, double-blind, comparison of filgrastim-sndz and the US-licensed reference biologic, filgrastim, in 218 patients with breast cancer being treated with docetaxel, doxorubicin, and cyclophosphamide (the TAC combination) chemotherapy for 6 cycles.32,33 Filgrastim-sndz or reference filgrastim was given at 5 mcg/kg/day from day 2 of each cycle until neutrophil recovery. The primary endpoint, the duration of severe neutropenia in cycle 1, was not significantly different between the two groups. Additionally, there were no differences in the rates of febrile neutropenia, days of fever, ANC nadir, and time to ANC recovery between the two products. Importantly, there was no difference in the rate of adverse effects related to treatment (cycle 1 — 20.6% biosimilar vs. 19.6% reference) and no neutralizing antibodies were detected.
Based on these data, the totality of evidence, and the mechanism of action being similar across indications, extrapolation was used to support FDA approval of filgrastim-sndz for all patent-eligible reference indications. Filgrastim-sndz is not considered interchangeable with filgrastim (Neupogen), as the pathway for this designation did not exist when the product was approved. Because filgrastim-sndz was developed and approved by FDA before adoption of the nonmemorable suffixes for biosimilars, it is the only product with a suffix based on the name of the developing company (Sandoz).
Filgrastim-aafi (Nivestym), manufactured by Pfizer, Inc., is another filgrastim biosimilar that gained FDA approval in July 2018.14 Filgrastim-aafi has also been approved for most of the indications of the reference product. Similar to filgrastim-sndz, filgrastim-aafi was based on a totality of the evidence, requiring demonstration of critical preclinical characteristics to demonstrate its highly similar nature to the reference biologic. Limited published data on filgrastim-aafi was available at the time this program was developed.
Tbo-filgrastim (Granix) is another G-CSF “follow-on” product that is approved by FDA to reduce the duration of severe neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia.34 However, Teva Oncology submitted a full Biologics License Application (BLA) prior to the introduction of the current FDA biosimilar approval pathway; thus, it is not technically a biosimilar or a generic product but a separately licensed biologic therapy.
A major consideration with biosimilar filgrastim products is the ability to dose the product for pediatric patients. Filgrastim-sndz and tbo-filgrastim are only available in prefilled syringes (300 mcg/0.5 mL and 480 mcg/0.8 mL). This hinders the ability to dose G-CSF in pediatric patients weighing less than 36 kg, as doses less than 0.3 mL cannot be accurately delivered. The reference biologic and filgrastim-aafi both come as prefilled syringes (300 and 480 mcg), as well as single-dose vials (300 and 480 mcg), making them more viable options in pediatric patients.
Biosimilar pegfilgrastim
Pegfilgrastim (Neulasta), the reference biologic, is a pegylated form of filgrastim. This pegylation increases the half-life of the white blood cell growth factor, enabling stimulation of granulocyte production for up to 14 days after a single dose.15 Pegfilgrastim is approved for adult and pediatric patients to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a clinically significant incidence of febrile neutropenia, and to increase survival in patients acutely exposed to myelosuppressive doses of radiation.
Pegfilgrastim-jmdb (Fulphila) and pegfilgrastim-cbqv (Udenyca) are FDA-approved pegfilgrastim biosimilars approved in fall 2018.3,16 Limited information was available on pegfilgrastim-cbqv when this program was prepared.21 Pegfilgrastim-jmdb showed high preclinical similarity to both the US-pegfilgrastim and EU-pegfilgrastim reference biological products through demonstration of similar myeloid (e.g., granulocyte) cell line proliferation and binding affinity at the G-CSF receptor. A phase 1, randomized, double-blind, three-way crossover trial in healthy volunteers was used to demonstrate similarity in PKs, PDs, and safety compared to reference pegfilgrastim. In this study, healthy volunteers were randomized to one of six possible treatment sequences and received 2 mg subcutaneously of either pegfilgrastim-jmdb (MYL-1401H), US-pegfilgrastim, or EU pegfilgrastim in various orders with 4-week washout periods between doses. The 2 mg dose was chosen rather than the usual 6 mg dose of reference pegfilgrastim as this dose is at the steepest portion of the dose-response curve and is thus most sensitive at detecting possible PD differences between the biosimilar and reference products.35 This study demonstrated highly a similar PK profile with pegfilgrastim-jmdb to the reference products, with nearly superimposable concentration-time profiles. Importantly, the primary PD parameters – ANC area under the curve (AUC) and ANC peak (Cmax) – were also highly similar among the three products. Other PD parameters, including CD34+ cell AUC and Cmax, were also highly similar. Regarding safety, there were no significant differences between adverse effects, treatment-emergent adverse effects, or severe adverse effects between the two products. There were also no differences in the rate of development of antidrug antibodies during therapy (22% pegfilgrastim-jmdb, 24% EU-pegfilgrastim, and 30% US-pegfilgrastim). Few patients overall developed neutralizing antibodies (as detected by a cell-based assay), and none who did had a significant change in ANC, loss of efficacy, or immune-related adverse effects.35
Based on the high degree of similarity of these pegfilgrastim biosimilars, EU-pegfilgrastim, and US-pegfilgrastim in preclinical characteristics and PK and PD in healthy volunteers, the FDA approved pegfilgrastim-jmdb for the same indication as reference pegfilgrastim but did not extrapolate this approval to patients acutely exposed to myelosuppressive doses of radiation because this indication has patent protection. As with other biosimilars, the pegfilgrastim biosimilars are not considered interchangeable.3,35
One technological development unique to the reference product pegfilgrastim has been the availability of the Neulasta Onpro on-body injector – a small, battery-powered, programmed drug delivery system that is applied to the patient’s skin after chemotherapy administration and delivers pegfilgrastim 24 hours after activation.36 It is recommended by National Comprehensive Cancer Network (NCCN) guidelines to administer white blood cell growth factors, including pegfilgrastim, at least 24 hours after chemotherapy, due to the theoretical risk of stimulating myeloid progenitor cells early after chemotherapy, exposing them to cytotoxic effects of chemotherapy and resulting in a more severe depth and duration of neutropenia.37,38 While this practice is controversial and the data are mixed, some studies have suggested enhanced myelosuppression when pegfilgrastim is given the same day as cytotoxic chemotherapy, rather than at least 24 hours following administration.37,39,40 Because of this, many insurance providers will not reimburse filgrastim or pegfilgrastim given on the same day as chemotherapy. The advantage of the on-body injector, which is only available with the reference pegfilgrastim, is that patients do not have to return to clinic the next day for pegfilgrastim. For patients in whom it is logistically challenging or inconvenient to return to clinic the next day, use of biosimilars of pegfilgrastim would be challenging, as they do not currently have the option of an on-body injector.
Biosimilar epoetin alfa
Epoetin alfa is an ESA that binds to erythroid progenitor cells and stimulates the production and differentiation of red blood cells (RBCs), similar to the endogenous hormone erythropoietin.41 Two identical epoetin alfa reference biological products (Procrit, Janssen; Epogen, Amgen) were approved under the same BLA; these have been marketed separately by the two companies pursuant to a product license agreement. Epoetin alfa is currently FDA approved to reduce allogeneic RBC transfusions in patients undergoing elective, noncardiac, nonvascular surgery, and in the treatment of anemia due to chronic kidney disease (CKD), in patients with anemia due to zidovudine in patients with HIV, and in patients with anemia due to concomitant myelosuppressive chemotherapy.24
In May 2018, epoetin alfa-epbx (Retacrit) became the first biosimilar ESA approved by FDA, and the action covered all indications of the reference product, epoetin alfa.42 Extensive preclinical analytical analyses determined epoetin alfa-epbx was highly similar to the reference product.41 The amino acid sequences of the two products are identical, and secondary and tertiary structures were shown to be highly similar via spectral methods. Overall, both products were highly similar from an analytic perspective, with few minor differences in posttranslational modifications of the protein. In addition, equivalency was demonstrated between epoetin alfa-epbx and the reference product with regards to in vivo and in vitro biopotency assays.
In addition to this extensive preclinical characterization demonstrating the highly similar nature of epoetin alfa-epbx, three clinical PK/PD studies have been conducted (EPOE-12-02, EPOE-14-01, and EPOE-10-08) as well as four clinical trials supporting the efficacy and safety of epoetin alfa-epbx (EPOE-10-13, EPOE-10-01, EPOE-11-04, and EPOE-11-03).41 Throughout all PK analyses, the confidence interval ratio for key PK parameters (e.g., Cmax, AUC) of epoetin alfa-epbx were within the predefined acceptance limits of 0.8–1.25, demonstrating the highly similar PK profile of the biosimilar product.41
The clinical studies supporting the similar safety and efficacy of epoetin alfa-epbx were conducted in patients with CKD on hemodialysis (HD) – one evaluated intravenous (IV) administration and the other evaluated subcutaneous (SQ) administration.41,43 This population was chosen because erythropoietin deficiency is the predominant cause of anemia in this population and patients with CKD on HD are the most erythropoietin-deficient population, thus providing the best sensitivity to detect differences in efficacy between the biosimilar and reference biologic. Furthermore, in contrast to cancer patients with anemia, CKD patients on HD are relatively immunocompetent, allowing for the assessment and comparison of immunogenicity between the two products.
Both studies enrolled adult patients on stable IV or SQ epoetin alfa (<600 units/kg/week) with stable hemoglobin (Hgb, mean between 9 and 11 g/dL) for 4 weeks prior to randomization. Patients who met entry criteria were randomized 1:1 to the reference biologic or epoetin alfa-epbx SQ (n = 246 total) or IV (n = 612 total) for 16 weeks or 24 weeks, respectively, with doses adjusted as needed to maintain Hgb in the 9 g/dL to 11 g/dL range. In both studies, the co-primary endpoints of mean weekly hemoglobin and mean weekly epoetin dose per kilogram of body weight during the last 4 weeks of treatment were within the predefined boundaries for equivalence between epoetin alfa-epbx and reference epoetin alfa. The safety analysis combined data from the aforementioned clinical studies using more than 800 patients and demonstrated no significant differences in the safety profiles between epoetin alfa-epbx and reference epoetin, including no differences in the rate of thromboembolic events (7.8% vs. 6.1%), hypertension (6.6% vs. 4.9%), potential allergic reactions (2.4% vs. 1.4%), myocardial infarction (0.9% vs. 0.7%), or cerebrovascular events (0.9% vs. 1.4%). There were also no cases of pure red cell aplasia (an immunogenic concern of ESAs), no differences in the rate of antidrug antibody formation (1% in both groups), and no difference in the rate of potential allergic reactions.41,43
Participants who completed 16–24 weeks of treatment were eligible to enroll in a long-term, 48-week, open-label study of epoetin alfa-epbx; it raised no additional safety concerns with the biosimilar product. Based on the high degree of similarity and similar mechanism across indications, FDA approval of epoetin alfa-epbx was extrapolated to all indications of the reference biologic; however, as with all other products approved thus far, the biosimilar is not considered interchangeable.41,43
Biosimilar bevacizumab-awwb
Bevacizumab (Avastin) is a humanized monoclonal IgG1 antibody against vascular endothelial growth factor (VEGF).17 VEGF normally binds to its receptors, stimulating proliferation of endothelial cells and new blood vessel formation, vital for continued cancer cell growth and proliferation. By binding VEGF and preventing this interaction, bevacizumab inhibits angiogenesis and effectively starves cancer cells of vital nutrients necessary for continued growth, spread, and metastasis. Bevacizumab is approved by FDA for a number of difficult-to-treat malignancies: Metastatic colorectal cancer; first-line for nonsquamous, nonsmall-cell lung cancer (NSCLC); progressive glioblastoma, metastatic renal cell carcinoma; persistent, recurrent, or metastatic cervical cancer; and epithelial ovarian, fallopian tube, or primary peritoneal cancer.
Bevacizumab-awwb (Mvasi) was the first biosimilar approved to treat cancer in September 2017. It was approved for all indications of the reference product, except epithelial ovarian, fallopian tube, or primary peritoneal cancer, as the reference product was not approved until June 2018 these indications and they are protected under orphan exclusivity.18,44 Bevacizumab-awwb is not considered an interchangeable biosimilar.
Bevacizumab-awwb was demonstrated to have a similar amino acid content (aside from a single-point mutation that results in a single amino acid difference that is not expected to have a structural or clinical impact) and a similar higher order structure to the US- and EU- reference product.45 Glycan analysis demonstrated bevacizumab-awwb has a higher fucose content and a higher mannose content. Increased fucosylation of glycan residues can decrease monoclonal antibody binding to Fc-gamma receptors on effector cells (e.g., NK cells, neutrophils, macrophages), thereby decreasing antibody-dependent cellular cytotoxicity (ADCC).46 However, because the primary mechanism of bevacizumab in the FDA-approved indications is to bind free VEGF and prevent receptor binding, reduced ADCC is not expected to have an impact on its clinical activity. High mannose content can result in increased clearance of monoclonal antibodies. Despite the higher mannose content, a 3-arm, randomized (1:1:1), parallel-group study in healthy volunteers demonstrated that 90% confidence intervals (CIs) for the ratios of the geometric means of AUC and Cmax were highly similar and within the 80%–125% prespecified limits for a 3 mg/kg single dose of US-reference bevacizumab, EU-reference bevacizumab, and bevacizumab-awwb.47
The highly similar analytical properties, preclinical characteristics, and PKs justified the undertaking of a randomized, double-blind comparative clinical study in patients with metastatic or recurrent nonsquamous NSCLC (Study 20120265).48 In this study, patients were randomized 1:1 to receive carboplatin/paclitaxel (an NCCN category 1 cytotoxic therapy option in this setting) plus either bevacizumab-awwb (n = 328) or EU-reference bevacizumab (n = 314) 15 mg/kg IV on day 1 of each 3 week cycle for 4 to 6 cycles. Bevacizumab-awwb or EU-reference bevacizumab was given as single-agent continuation maintenance until progression or intolerable toxicity. The primary endpoint of overall response rate was 39% in the bevacizumab-awwb group and 41.7% in the EU-reference bevacizumab group (risk ratio 0.93, 90% CI 0.8–1.09), which was well within the predefined similarity margins proposed by Amgen (similarity margin of 0.67 to 1.5) and FDA (equivalence margin of 0.73 to 1.36). Secondary endpoints of response duration and progression-free survival were also similar between the two groups (Table 2). The toxicity profile of the two products was highly similar between bevacizumab-awwb and EU-reference bevacizumab, including VEGF-specific adverse effects. Importantly, there were no differences in the immunogenicity between bevacizumab-awwb and EU-reference bevacizumab. The rate of treatment-emergent antidrug antibodies was low (1.4% vs. 2.5%, respectively) and had no impact on safety, efficacy, or PKs.48
| Table 2. Bevacizumab-awwb Clinical Outcomes and Adverse Events (Study 20120265) |
| |
Bevacizumab-awwb (n = 328)
N (%) |
EU-bevacizumab (n = 314)
N (%) |
Overall response rate
Complete response
Partial response
Stable disease |
128 (39%)
2 (0.6%)
126 (38.4%)
144 (43.9%) |
131 (41.7%)
2 (0.6%)
129 (41.1%)
137 (43.6%) |
Duration of response, median |
5.8 months |
5.6 months |
Progression-free survival, median |
6.6 months |
7.9 months |
| |
Hypertension
Any grade
Grade 3/4 |
19%
7% |
16%
6% |
Hemorrhages
Any grade
Grade 3/4 |
22%
2% |
21%
1% |
Venous thromboembolism
Any grade
Grade 3/4 |
4%
2% |
5%
3% |
Arterial embolism
Any grade
Grade 3/4 |
2%
1% |
1%
1% |
Proteinuria
Any grade
Grade 3/4 |
8%
<1% |
6%
<1% |
| Source: Reference 48. |
Compared with supportive care biosimilars in which the mechanism of action is typically the same across indications, the extrapolation of biosimilars used to treat cancer to other indications requires careful consideration. In the case of bevacizumab-awwb, the mechanism of action is consistent across indications – the neutralization of VEGF, preventing binding to its receptors, thus inhibiting angiogenesis and tumor growth. Additionally, the PK properties across various FDA-approved doses are consistent. Because PK similarity between bevacizumab-awwb and reference bevacizumab was demonstrated at a 3 mg/kg dose in healthy volunteers, as well as in the NSCLC study when given at 15 mg/kg, a similar PK profile can be inferred for other FDA-approved indications. Although some VEGF-related toxicities are more common in certain indications due to the nature of the malignancies (e.g., a higher rate of hemoptysis in NSCLC studies), the types of adverse effects are similar across indications. Because there were no safety signals in the comparative clinical study, a similar adverse effect profile of bevacizumab-awwb can be expected in other indications. Overall, based on the common mechanism of action and extensive characterization of bevacizumab-awwb, FDA supported the conclusion that extrapolation of approval to all indications of the reference product (with the exception of epithelial ovarian, fallopian tube, or primary peritoneal cancer as noted above) be granted.
Biosimilar trastuzumab-dkst
Trastuzumab (Herceptin) is a humanized IgG1 kappa monoclonal antibody that selectively binds to the extracellular domain of the human epidermal growth factor receptor 2 protein (HER2).19 HER2 is a transmembrane tyrosine kinase that regulates cellular proliferation, adhesion, and differentiation. It is expressed in 20% to 30% of breast and gastric cancers.49,50 Trastuzumab binding to HER2 results in inhibition of dimerization important for downstream activation and survival signaling, increased destruction of the HER2 receptor, decreased shedding of the extracellular portion of the HER2 receptor, and, importantly, initiation of ADCC. Trastuzumab is currently approved by FDA for the treatment of HER2-overexpressing (HER2+) breast cancer in the adjuvant or metastatic setting, as well as in the treatment of HER2+ metastatic gastric or gastroesophageal junction adenocarcinoma.19 Trastuzumab-dkst (Ogivri) was approved in December 2017 as the first biosimilar trastuzumab product approved and was approved for the same indications as the reference biologic. As with other currently approved biosimilars in the US, it is not considered interchangeable.20
Analytically, trastuzumab-dkst was found to be highly similar to the reference product. Its amino acid sequence is identical with a similar higher order structure.51 There were minor differences in the charge profiles of trastuzumab-dkst compared with US- and EU-reference trastuzumab, as well as a higher mannose content, higher sialic acid content, and a lower percentage of nonglycosylated heavy chain. Despite the differences between the agents in posttranslational modification, trastuzumab-dkst demonstrated equivalent HER2 binding in vitro, equivalent inhibition of proliferation in a HER2+ breast cancer cell line, and equivalent ADCC activity in the aforementioned cell line. Similarly, the PKs of trastuzumab-dkst were found to be highly similar to US-reference and EU-reference trastuzumab in a double-blind, randomized, parallel group, phase 1 study in healthy adult male volunteers.52 In this study, patients were given a 8 mg/kg dose of one of the three products over 90 minutes; the concentration-time profiles of the biological products were nearly superimposable, and the AUC and Cmax were well within the 80% to 125% prespecified similarity margin.
| Figure 2. Trastuzumab-dkst Clinical Study Schema
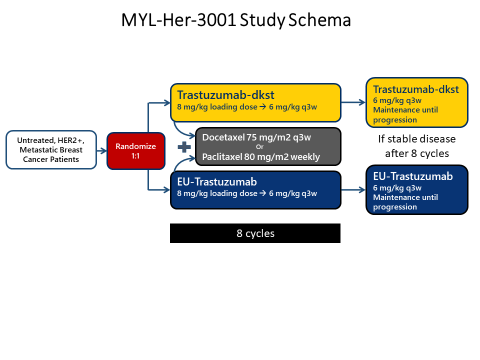
|
| Source: Reference 51. |
From a clinical perspective, trastuzumab-dkst was studied in a double-blind, randomized, parallel group study in patients with untreated, HER2-positive, metastatic breast cancer (n = 458) (Figure 2).51 Patients were randomized 1:1 to trastuzumab-dkst or EU-reference trastuzumab in combination with either paclitaxel or docetaxel for a minimum of 8 cycles. Responding patients with at least stable disease after this portion of the study were offered continuance on single-agent trastuzumab-dkst or EU-reference trastuzumab. Trastuzumab-dkst and reference biologic were given as an 8 mg/kg IV loading dose, then 6 mg/kg every 3 weeks thereafter. The primary endpoint of overall response rate was no different between trastuzumab-dkst and the reference biologic [70% vs. 64%, ORR 1.09 (90% CI; 0.98–1.22)], which fell between the prespecified equivalence margin. Median progression-free survival (PFS) and overall survival (OS) were also not significantly different between the two products.
The side effect profile of both agents was also highly similar – there were no differences in the rates of treatment-emergent adverse effects (TEAEs), grade 3 or higher TEAEs, or events leading to discontinuation of study drug. Of special interest to trastuzumab, there were no significant differences in the cardiotoxicity profile of the two agents, although these events were numerically higher in the trastuzumab-dkst arm (cardiac failure 2.4% vs. 0.8%; Figure 3). The majority of cardiac adverse events were grade 1 and 2 in nature and resolved with time. The immunogenicity of both products was low and no different between the two products. Based on the highly similar preclinical characterization, the consistent mechanism of action across indications, similar PK profiles, and a highly similar safety and efficacy profile in metastatic breast cancer, the FDA supported approval of the trastuzumab-dkst biosimilar for all indications of the reference product.51
| Figure 3. Key Response Rates and Outcomes with Trastuzumab-dkst
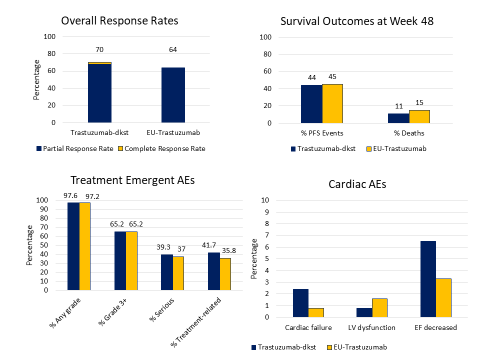
|
| Source: Reference 51. |
Case Study 2
A patient receiving myelosuppressive chemotherapy for cancer requires supportive therapy for preventing neutropenia. Her physician has ordered Neulasta Onpro, a pegfilgrastim product with an on-body injector. This dosage form is needed in this case since the patient lives 3 hours from the treatment center, and the patient cannot return the following day for administration of the dose because of both cost and lack of transportation. However, the patient’s managed care plan does not cover this product, and the cost to the patient is prohibitive. What factors can be presented in seeking approval for this product from the patient’s managed care plan?
As discussed above, white blood cell growth factors, including pegfilgrastim, should not be given for at least 24 hours after chemotherapy because of the theoretical risk of stimulating myeloid progenitor cells early after chemotherapy, which exposes them to cytotoxic effects of chemotherapy and results in a more severe depth and duration of neutropenia. In fact, many insurance providers will not reimburse filgrastim or pegfilgrastim given on the same day as chemotherapy, a fact that can be discussed with the patient’s plan. Since the patient is simply not able to return to the clinic for a dose the next day and cannot afford the on-body device, options include having the dose administered by a health professional convenient to the patient, teaching the patient or a caregiver to administer the dose, or using the on-body device. |
Oncology Biosimilars in Late Clinical Development
Biosimilar rituximab
Rituximab is a chimeric monoclonal antibody directed against the CD20 antigen present on the surface of pre-B and mature-B lymphocytes.53 Upon binding the CD20 on the cell surface, rituximab mediates cell death via ADCC, complement-dependent cytotoxicity, and direct induction of cell death via apoptosis. Rituximab is approved by FDA for the treatment of several disorders in which cells of the B-lineage are pathogenic — non-Hodgkin’s lymphoma (NHL), chronic lymphocytic leukemia (CLL), rheumatoid arthritis (RA), Wegener’s granulomatosis, microscopic polyangiitis, and pemphigus vulgaris.54 While several rituximab biosimilars are approved and being used clinically in Europe (Truxima, Blitzima, Ritemvia, and Rituzena marketed by Celltrion; Riximyo marketed by Sandoz), there has yet to be a rituximab biosimilar approved in the US.55
A biosimilar rituximab approved in the EU and three countries, GP2013 (Sandoz), was in development for US marketing, but after FDA twice requested additional information, the company chose not to pursue the application any further. A BLA for the product was originally accepted by FDA for review in September 2017. Along with extensive preclinical and analytical characterization, clinical studies supporting this BLA were the ASSIST-RA and ASSIST-FL studies in patients with RA and follicular lymphoma (FL, a type of non-Hodgkin lymphoma), respectively. The ASSIST-RA study was a randomized, double-blind, parallel group study in patients with active RA despite prior tumor necrosis factor inhibitor therapy (n = 312).56 The primary PK endpoint of AUC was similar between biosimilar rituximab and EU-reference or US-reference rituximab. Additionally, the main PD objective, peripheral B cell depletion, was equivalent among the three products. Safety and efficacy of the two products were also similar, as assessed by C-reactive protein at week 24, as well as numerous other response measures (both clinician- and patient-reported outcomes).
ASSIST-FL was a multinational, double-blind, randomized controlled trial of adult patients with previously untreated, Ann Arbor stage III/IV FL patients who were randomized 1:1 to receive CVP (cyclophosphamide, vincristine, prednisone), plus either biosimilar rituximab (GP2013) or EU-reference rituximab.57 Responding patients were offered maintenance therapy for up to 2 years with either product (double-blinded). The primary endpoint, the centrally reviewed overall response rate, was statistically equivalent between GP2013 and reference rituximab (87% vs. 88%, respectively; difference -0.4%, 95% CI –5.94 to 5.14%). There were no differences in the adverse effect profile of the two agents, with rates was low and not different between the two groups. Estimated investigator-assessed median PFS was not reached in either group; however, the proportion of patients with progression events was statistically higher in the GP2013 group compared with reference rituximab (30% vs. 24%, HR 1.31, 90% CI 1.02–1.69). It is important to note that more than 70% of patients in both groups were censored, and there was no central review of progression events. Nonetheless, the differences in PFS raise concern. Another concern with the data is the use of CVP as the chemotherapy backbone. While R-CVP is an appropriate regimen for some patients, in the US, R-bendamustine and R-CHOP are preferred by many clinicians because some data suggest greater efficacy of these treatment approaches.58–61
In May 2018, the company said it had received a Complete Response Letter for the GP2013 biosimilar and would continue discussions with FDA about the product.62 In November 2018, Sandoz announced that after receiving and reviewing additional data on GP2013, FDA asked for more information, and that by the time the company could satisfy this requirement, it expected that “the patient and marketplace needs in the US will be satisfied.” Thus, the company elected not to pursue marketing of GP2013 in the US.63
CT-P10 is biosimilar rituximab being developed by Celltrion that is in late clinical development.64 Along with highly similar analytic properties, CT-P10 was shown to have similar PK, PD, immunogenicity, efficacy, and safety in patients with RA, compared with US- and EU-reference rituximab.65 CT-P10 was also studied in a randomized, double-blind, parallel-group phase 3 controlled trial in patients with Ann Arbor stage III/IV untreated FL (n = 140). Patients were randomized 1:1 to CVP plus either CT-P10 (biosimilar rituximab) or reference rituximab (EU- or US-reference, depending on the treating site); responding patients received maintenance CT-P10 or reference rituximab for up to 2 years. The published interim results of the study (patients receiving up to week 24 of therapy) demonstrated similar overall response rates (97% CT-P10 vs. 92.6% reference rituximab), similar B cell depletion kinetics, PK parameters within the 80% to 125% bioequivalence margin, and a similar adverse effect profile in the two groups. Longer-term data are required for assessment of other secondary endpoints, including PFS and OS.66 FDA has submitted a Complete Response Letter to Celltrion requesting supplementary information, thus initially rejecting the BLA for this rituximab biosimilar.67
Biosimilar cetuximab
Cetuximab (Erbitux) is a chimeric monoclonal antibody directed against the extracellular domain of the human epidermal growth factor receptor (EGFR).68 By binding EGFR on tumor cells, cetuximab prevents binding of epidermal growth factor and other ligands, thus preventing downstream activation of kinases essential for cell growth and VEGF production. Binding of cetuximab to EGFR also elicits ADCC against certain tumor types.
Cetuximab is currently approved by FDA for head and neck cancer and colorectal cancer.69 Several cetuximab biosimilars are in clinical development, with a biosimilar/“biobetter” from MabTech-Sorrento – STI-001 – the furthest along in development. A “biobetter” is similar to a biosimilar, except that superiority over the reference biologic is sought in at least one aspect of the biobetter’s clinical profile, through various modifications of structure or formulation, in an effort to improve upon the safety or efficacy of the reference biologic.70 A confirmatory, randomized controlled trial of STI-001 in EGFR-expressing metastatic colorectal cancer was conducted, comparing STI-001 with irinotecan vs. irinotecan alone (n = 501).71 The combination showed a similar improvement in overall response rate (32.6% vs. 12.8%) and PFS (median 5.6 vs. 3.2 months) to that seen in trials assessing the addition of reference cetuximab to irinotecan chemotherapy.71,72 Differences in the production method of STI-001 vs. reference cetuximab are thought to produce differences in posttranslational modifications that may be responsible for a different rate of hypersensitivity reactions with the two products. To illustrate, in previous studies with the reference biologic, more than 10% of patients demonstrated grade 3/4 hypersensitivity reactions, compared with no patients in the phase 3 study of STI-001, although results have not been published to date.71
Conclusion
Biologic therapies have become a double-edged sword in oncology treatment – while they have revolutionized cancer therapy and improved outcomes for patients, they have contributed to the significant rise in healthcare costs in the treatment of patients with cancer. Biosimilar commercial development and marketing has lagged in the US compared with the EU for a variety of legal, regulatory, and clinical reasons. Six biosimilar agents are now available, and several agents are in late clinical development. These biosimilar therapies have the potential to significantly reduce costs and improve access to care. Because biosimilar approval follows a unique pathway for approval, it is critical for pharmacists to understand the data involved in bringing these therapies to market so that appropriate formulary decisions can be made.
References
- Lyman GH, Zon R, Harvey RD, Schilsky RL. Rationale, opportunities, and reality of biosimilar medications. N Engl J Med. 2018;378(21):2036-2044. doi:10.1056/NEJMhle1800125.
- IBM Watson Health. Micromedex (electronic version). http://truvenhealth.com/Products/Micromedex. Accessed October 5, 2018.
- U.S. Food & Drug Administration. Biosimilars — Biosimilar and Interchangeable Products. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580419.htm. Accessed September 24, 2018.
- US Food and Drug Administration. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry.; 2015. https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf. Accessed October 5, 2018.
- US Food and Drug Administration. Considerations in Demonstrating Interchangeability With a Reference Product: Guidance for Industry - Draft Guidance. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed October 5, 2018.
- Cohen HP, Blauvelt A, Rifkin RM, et al. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463-478. doi:10.1007/s40265-018-0881-y.
- National Conference of State Legislatures. State Laws and Legislation Related to Biologic Medications and Substitution of Biosimilars. http://www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx. Accessed October 2, 2018.
- Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Ureña S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76(2):346-354. doi:10.1136/annrheumdis-2015-208783.
- Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76(2):355-363. doi:10.1136/annrheumdis-2015-208786.
- Press Announcements - FDA approves Inflectra, a biosimilar to Remicade. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Accessed October 5, 2018.
- US Food and Drug Administration. Nonproprietary Naming of Biological Products: Guidance for Industry; 2017. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Accessed October 5, 2018.
- Stevenson JG, Green L. Biologics, pharmacovigilance, and patient safety: it’s all in the name. J Manag Care Spec Pharm. 2016;22(8):927-930. doi:10.18553/jmcp.2016.22.8.927.
- US Food and Drug Administration. Biosimilar Product Information; 2018. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580432.htm. Accessed November 13, 2018.
- Pfizer. Filgrastim-Aafi (Nivestym) Prescribing Information. Lake Forest, IL. www.fda.gov/medwatch. Accessed October 5, 2018.
- Amgen. Neulasta (Pegfilgrastim) Prescribing Information. Thousand Oaks, CA. www.fda.gov/medwatch. Accessed October 5, 2018.
- Mylan. Fulphila (Pegfilgrastim-Jmdb) Prescribing Information. Zurich, Switzerland. www.fda.gov/medwatch. Accessed October 5, 2018.
- Genentech. Avastin (bevacizumab) Prescribing Information. South San Francisco, CA. www.fda.gov/medwatch. Accessed October 5, 2018.
- Amgen. MVASI (bevacizumab-awwb) Prescribing Information. Thousand Oaks, CA. www.fda.gov/medwatch. Accessed October 5, 2018.
- Genentech. Herceptin (trastuzumab) Prescribing Information. South San Francisco, CA. www.fda.gov/medwatch. Accessed October 5, 2018.
- Mylan. Ogivri (trastuzumab-dkst) Prescribing Information. Zurich, Switzerland. www.fda.gov/medwatch. Accessed October 5, 2018.
- Coherus Biosciences. Pegfilgrastim-cbqv (Udenyca) Prescribing Information. Redwood City, CA. www.accessdata.fda.gov/drugsatfda_docs/label/2018/761039s000lbl.pdf. Accessed November 13, 2018.
- Amgen. Filgrastim (Neupogen) Prescribing Information. Thousand Oaks, CA. www.fda.gov/medwatch. Accessed October 5, 2018.
- FDA ODAC Document. Zarxio clinical review application type original 351(k). https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM512508.pdf. Accessed October 5, 2018.
- Amgen. Epogen (epoetin alfa) Prescribing Information. Thousand Oaks, CA. www.fda.gov/medwatch. Accessed October 5, 2018.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Pharm. 2013;70(22):2004-2017. doi:10.2146/ajhp130119.
- Grewal S, Ramsey S, Balu S, Carlson JJ. Cost-savings for biosimilars in the United States: a theoretical framework and budget impact case study application using filgrastim. Expert Rev Pharmacoecon Outcomes Res. 2018;18(4):447-454. doi:10.1080/14737167.2018.1476142.
- Moorkens E, Vulto AG, Huys I, Dylst P, Godman B, Keuerleber S, et al. Policies for biosimilar uptake in Europe: an overview. Bochenek T, ed. PLoS One. 2017;12(12):e0190147. doi:10.1371/journal.pone.0190147.
- Goldsmith D, Dellanna F, Schiestl M, Krendyukov A, Combe C. Epoetin biosimilars in the treatment of renal anemia: what have we learned from a decade of European experience? Clin Drug Investig. 2018;38(6):481-490. doi:10.1007/s40261-018-0637-1.
- Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3. http://www.ncbi.nlm.nih.gov/pubmed/30083415. Accessed October 5, 2018.
- US Food and Drug Administration. FDA approves Neupogen for treatment of patients with radiation-induced myelosuppression following a radiological/nuclear incident. https://www.fda.gov/emergencypreparedness/counterterrorism/medicalcountermeasures/aboutmcmi/ucm443245.htm. Accessed November 21, 2018.
- Holzmann J, Balser S, Windisch J. Totality of the evidence at work: the first U.S. biosimilar. Expert Opin Biol Ther. 2016;16(2):137-142. doi:10.1517/14712598.2016.1128410.
- Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953. doi:10.1093/annonc/mdv281.
- Blackwell K, Gascon P, Krendyukov A, et al. Safety and efficacy of alternating treatment with EP2006, a filgrastim biosimilar, and reference filgrastim: a phase III, randomised, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2018;29(1):244-249. doi:10.1093/annonc/mdx638.
- Elayan MM, Horowitz JG, Magraner JM, Shaughnessy PJ, Bachier C. Tbo-filgrastim versus filgrastim during mobilization and neutrophil engraftment for autologous stem cell transplantation. Biol Blood Marrow Transpl. 2015;21(11):1921-1925. doi:10.1016/j.bbmt.2015.05.024.
- Waller CF, Tiessen RG, Lawrence TE, et al. A pharmacokinetics and pharmacodynamics equivalence trial of the proposed pegfilgrastim biosimilar, MYL-1401H, versus reference pegfilgrastim. J Cancer Res Clin Oncol. 2018;144(6):1087-1095. doi:10.1007/s00432-018-2643-3.
- Yang B-B, Morrow PK, Wu X, et al. Comparison of pharmacokinetics and safety of pegfilgrastim administered by two delivery methods: on-body injector and manual injection with a prefilled syringe. Cancer Chemother Pharmacol. 2015;75(6):1199-1206. doi:10.1007/s00280-015-2731-x.
- Meropol NJ, Miller LL, Korn EL, et al. Severe myelosuppression resulting from concurrent administration of granulocyte colony-stimulating factor and cytotoxic chemotherapy. J Natl Cancer Inst. 1992;84(15):1201-1203. http://www.ncbi.nlm.nih.gov/pubmed/1378905. Accessed September 26, 2018.
- Sue Becker P, Alwan L, Brown A, et al. NCCN Guidelines Version 2.2018 Myeloid Growth Factors; 2018. https://www.nccn.org/professionals/physician_gls/pdf/myeloid_growth.pdf. Accessed September 26, 2018.
- Burris HA, Belani CP, Kaufman PA, et al. Pegfilgrastim on the same day versus next day of chemotherapy in patients with breast cancer, non–small-cell lung cancer, ovarian cancer, and non-Hodgkin’s lymphoma: results of four multicenter, double-blind, randomized phase II studies. J Oncol Pract. 2010;6(3):133-140. doi:10.1200/JOP.091094.
- Lyman GH, Allcott K, Garcia J, et al. The effectiveness and safety of same-day versus next-day administration of long-acting granulocyte colony-stimulating factors for the prophylaxis of chemotherapy-induced neutropenia: a systematic review. Support Care Cancer. 2017;25(8):2619-2629. doi:10.1007/s00520-017-3703-y.
- Epoetin Hospira A Proposed Biosimilar to Epogen/Procrit (Epoetin Alfa) Briefing Document for the Oncologic Drugs Advisory Committee; 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559968.pdf. Accessed October 5, 2018.
- Thadhani R, Guilatco R, Hymes J, et al. Switching from epoetin alfa (Epogen®) to epoetin alfa-epbx (RetacritTM) using a specified dosing algorithm: a randomized, non-inferiority study in adults on hemodialysis. Am J Nephrol. 2018;48(3):214-224. doi:10.1159/000492621.
- Fishbane S, Singh B, Kumbhat S, Wisemandle WA, Martin NE. Intravenous epoetin alfa-epbx versus epoetin alfa for treatment of anemia in end-stage kidney disease. Clin J Am Soc Nephrol. 2018;13(8):1204-1214. doi:10.2215/CJN.11631017.
- Center for Drug Evaluation and Research. Approved drugs — FDA Approves Bevacizumab In Combination With Chemotherapy for Ovarian Cancer. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610664.htm. Accessed October 5, 2018.
- Amgen. ABP215 FDA Briefing Document Oncologic Drugs Advisory Committee; 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM566365.pdf. Accessed October 5, 2018.
- Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115(22):4393-4402. doi:10.1182/blood-2009-06-225979.
- Markus R, Chow V, Pan Z, Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother Pharmacol. 2017;80(4):755-763. doi:10.1007/s00280-017-3416-4.
- Casak SJ, Lemery SJ, Chung J, et al. FDA’s approval of the first biosimilar to bevacizumab. Clin Cancer Res. 2018;24(18):4365-4370. doi:10.1158/1078-0432.CCR-18-0566.
- Cronin KA, Harlan LC, Dodd KW, et al. Population-based estimate of the prevalence of HER-2 positive breast cancer tumors for early stage patients in the US. Cancer Invest. 2010;28(9):963-968. doi:10.3109/07357907.2010.496759.
- Boku N. HER2-positive gastric cancer. Gastric Cancer. 2014;17(1):1-12. doi:10.1007/s10120-013-0252-z.
- MYL-1401O FDA Briefing Document Oncologic Drugs Advisory Committee Meeting; 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM566369.pdf. Accessed October 5, 2018.
- Waller CF, Vutikullird A, Lawrence TE, et al. A pharmacokinetics phase 1 bioequivalence study of the trastuzumab biosimilar MYL-1401O vs. EU-trastuzumab and US-trastuzumab. Br J Clin Pharmacol. 2018;84(10):2336-2343. doi:10.1111/bcp.13689.
- Feugier P. A review of rituximab, the first anti-CD20 monoclonal antibody used in the treatment of B non-Hodgkin’s lymphomas. Futur Oncol. 2015;11(9):1327-1342. doi:10.2217/fon.15.57.
- Genentech/Biogen. Rituxan (rituxumab) Prescribing Information. Accessed November 13, 2018.
- Santos SB, Sousa Lobo JM, Silva AC. Biosimilar medicines used for cancer therapy in Europe: a review. Drug Discov Today. September 2018. doi:10.1016/j.drudis.2018.09.011.
- Smolen JS, Cohen SB, Tony H-P, et al. A randomised, double-blind trial to demonstrate bioequivalence of GP2013 and reference rituximab combined with methotrexate in patients with active rheumatoid arthritis. Ann Rheum Dis. 2017;76(9):1598-1602. doi:10.1136/annrheumdis-2017-211281.
- Jurczak W, Moreira I, Kanakasetty GB, et al. Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol. 2017;4(8):e350-e361. doi:10.1016/S2352-3026(17)30106-0.
- Siddhartha G, Vijay P. R-CHOP versus R-CVP in the treatment of follicular lymphoma: a meta-analysis and critical appraisal of current literature. J Hematol Oncol. 2009;2:14. doi:10.1186/1756-8722-2-14.
- Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203-1210. doi:10.1016/S0140-6736(12)61763-2.
- Flinn IW, van der Jagt R, Kahl BS, Wood P, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123(19):2944-2952. doi:10.1182/blood-2013-11-531327.
- Luminari S, Ferrari A, Manni M, et al. Long-term results of the FOLL05 trial comparing R-CVP Versus R-CHOP versus R-FM for the initial treatment of patients with advanced-stage symptomatic follicular lymphoma. J Clin Oncol. 2018;36(7):689-696. doi:10.1200/JCO.2017.74.1652.
- Davio K. FDA Rejects Sandoz’s Proposed Rituximab Biosimilar. https://www.centerforbiosimilars.com/news/fda-rejects-sandozs-proposed-rituximab-biosimilar. Accessed October 5, 2018.
- Davio K. Sandoz Abandons its US Biosimilar Rituximab Plans. https://www.centerforbiosimilars.com/news/sandoz-abandons-its-us-biosimilar-rituximab-plans. Accessed November 7, 2018.
- Suh C-H, Kasay AB, El-Khouri EC, et al. Pharmacokinetics and safety of three formulations of rituximab (CT-P10, US-sourced Innovator Rituximab and EU-sourced Innovator Rituximab) in patients with rheumatoid arthritis: results from phase 3 randomized controlled trial over 24 weeks — ACR meeting abstracts. Am Coll Rheumatol Annu Meet. 2016. Abstract 1634. https://acrabstracts.org/abstract/pharmacokinetics-and-safety-of-three-formulations-of-rituximab-ct-p10-us-sourced-innovator-rituximab-and-eu-sourced-innovator-rituximab-in-patients-with-rheumatoid-arthritis-results-from-phase-3-r/. Accessed October 5, 2018.
- Yoo DH, Suh C-H, Shim SC, et al. A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(3):566-570. doi:10.1136/annrheumdis-2016-209540.
- Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Hematol. 2017;4(8):e362–e373.
- Davio K. FDA Rejects Celltrion’s Rituximab and Trastuzumab Biosimilars. https://www.centerforbiosimilars.com/news/fda-rejects-celltrions-rituximab-and-trastuzumab-biosimilars. Accessed October 5, 2018.
- Kirkpatrick P, Graham J, Muhsin M. Cetuximab. Nat Rev Drug Discov. 2004;3(7):549-550. doi:10.1038/nrd1445.
- Bristol-Myers Squibb/Lilly. Erbitux (cetuximba) Prescribing Information. 2012. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125084s0228lbl.pdf Accessed November 13, 2018.
- Dolinar RO, Reilly MS. The future of biological therapy a pathway forward for biosimilars. Generics Biosimilars Initiative J. 2013;2(1):36-40. doi:10.5639/gabij.2013.0201.014.
- Sorrento Therapeutics. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Accessed October 5, 2018.
- Sobrero AF, Maurel J, Fehrenbacher L, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(14):2311-2319. doi:10.1200/JCO.2007.13.1193.
Back to Top