
Expired activity
Please go to the PowerPak
homepage and select a course.
Examining Real-World Evidence in the Management of Breast Cancer: Pharmacist Implications
INTRODUCTION
Real-world evidence (RWE) is evidence that is gained about the uses and potential benefits or risks of a medical product that is derived from data directly relating to patient health status and/or the delivery of healthcare (i.e., real-world data [RWD]).1-3 RWE has been collected for decades, but its applications are just now growing at a rapid pace. The continuous expansion of digital RWD has been followed by an increase in analysis and generation of RWE. Healthcare providers, researchers, businesses, and government agencies are all interested in RWE, as it is a useful tool to close persistent evidence gaps.4
All vested healthcare parties want to understand how drugs are used in the real world and the outcomes they produce. Despite well-executed randomized clinical trials, many patients in the “real world” are treated differently than those in the original registry trial that led to a drug’s approval by the United States (U.S.) Food and Drug Administration (FDA). Additionally, rare adverse events that were not detected in the clinical trials may occur during real-world use. This RWD can be evaluated to fill the gaps that exist in these settings.4
The focuses and types of analyses performed with RWD reflect the work of the researcher. For instance, the FDA is interested in the safe and effective use of medications and it has an entire division devoted to pharmacovigilance that monitors and investigates reports of drug-related adverse events and, ultimately, communicates safety concerns. The fruit of this labor can be seen in the safety sections of package inserts. Similarly, pharmaceutical companies desire to understand how their medications are being used and, potentially, misused. Marketing teams uses this information to identify barrier to drug utilization and develop plans to overcome these barriers. They are also interested in identifying and preventing any practices that could lead to negative outcomes for which they may have legal accountability. Finally, academic practitioners utilize RWE to help craft practice guidelines in an effort to standardize treatment and optimize outcomes.1-4
There are many sources of RWD (Table 1),2 but data collection actually begins when a physician applies his or her impression of the clinical trial data to an actual patient. This extrapolation of clinical trial data is necessary because many patients are not eligible for clinical trials or choose not to participate. Not every patient situation nor every doctor’s decision is the same, but, given the vast amount of RWD and its accessibility for extraction and analysis, the scientific conclusions gained from the real-world use of treatments are potentially more valid now than ever before. In order to expand the utility of RWE, standardization and articulated thresholds of expectations must be created and embraced by regulatory agencies. This was the basis for the amendment to the Food, Drug, and Cosmetics (FD&C) Act known as the 21st Century Cures Act.5
| Table 1. Sources of Real-World Data2 |
- Electronic health records
- Claims and billing activities
- Product and disease registries
- Patient-generated data, including in home-use settings
- Data gathered from other sources that can inform on health status, such as mobile devices
|
THE 21ST CENTURY CURES ACT
On December 13, 2016, the 21st Century Cures Act was signed into law. The stated goal for this amendment to the FD&C Act is “to help accelerate medical product development and bring new innovations and advances to patients who need them faster and more efficiently.”5 Because this is a law, it requires accountability with reporting to congress. Title III subtitle C applies to RWD and RWE: it includes “Novel Clinical Trial Designs” (Sec. 3021) and “Real World Evidence” (Sec. 3022).6 Each section includes the type of deliverable, the statutory deadline, the responsible organization, the date completed, and access to the results.5,7 There was a public meeting in June 2018 to discuss novel clinical trial designs that was facilitated by the FDA’s Center for Drug Evaluation and Research (CDER), and the CDER’s final guidance and framework for the RWE program was published in December 2018.3
Study designs
The strength of a scientific trial is directly linked to the study design. The gold standard, and the strongest design, in drug development is a randomized, controlled clinical trial (RCT). The hierarchy of the strengths of study designs is depicted in Figure 1.8 It could be argued that a meta-analysis of RCTs is stronger than a single RCT, but this module will not address that debate; instead, it will primarily focus on RWE methodologies in which treatment is not assigned by a protocol.
In part, the 21st Century Cures Act aims to impact RCT design to make the process more efficient.3-5 Specifically, the CDER is working to establish standardized quality standards for adaptive study designs. These designs tend to be complex and can alter the study significantly while it is accruing patients. The essence of an adaptive study design is to provide ongoing analysis to predict the probability of one arm being superior to another arm or one group of patients performing better or worse than another group and adjusting the protocol as it moves forward. Consequently, the number of subjects accrued into each arm can vary; the treatment allocation and even the patient population can change during the trial. Philosophically, it makes sense to determine the likelihood that one arm is better than another or that one identifiable patient group will perform better than another and preferentially assign subjects to the treatment arm that will be best. In the end, such an approach minimizes the number of patients receiving therapy with less effective or more toxic treatment. The challenge is to make these changes in a scientifically valid fashion. The idea of using Bayesian methods to adapt a clinical trial has been discussed in the literature since the early 1990’s. However, few, if any, registry drug trials have been performed with this study design methodology. This will likely change with the 21st Century Cures Act, particularly since the FDA is responsible for developing the plan and expectations in this area.
Figure 1. The Comparative Strengths of Study Designs8
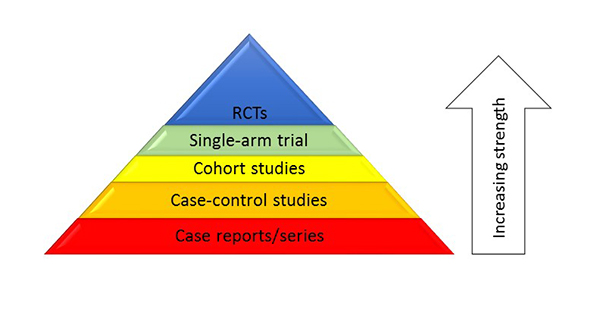 |
| RCT, randomized, controlled clinical trial. |
The differences between clinical trials and studies evaluating RWE are outlined in Table 2.8 The single biggest difference between them is the personalization of treatment in the real world and adherence to a strict protocol for all patients in a trial that assigns treatment. Physicians are dedicated to caring for patients who have a broad spectrum of issues that can impact clinical decisions. As such, physicians may be described as “voting with their feet,” meaning that they extrapolate evidence from an RCT to a treatment plan they believe is best for a specific patient: they may select different drug dosing regimens, patient follow-up plans, or durations of therapy, among many other deviations from the clinical trial protocol. The tighter the inclusion and exclusion criteria of an RCT, the bigger the evidence gaps between the published trial results and most patients in a real-world setting. These real-world patients can be referred to as “non-protocolized” patients. The evidence gaps between these scenarios can be addressed with analyses of how physicians “vote with their feet.”
| Table 2. Differences between Clinical Trials and Real-World Evidence Studies8 |
| Clinical trials |
Real-world evidence studies |
- Robust control of patient population
- Strict inclusion/exclusion criteria
- Strict guidance on drug dosing
- Starting dose
- Dose-adjustment criteria
- Control of timing of evaluations
- Radiologic scans
- Definitions of response
- Defined criteria for “off study”
- Progressive disease
- Toxicity
- Duration of therapy
|
- Epidemiology approach is most common
- Statistically controlled populations
- Typically include very large numbers of patients at the cost of limited data quality (particularly secondary data)
- Useful for uncommon events
- Rare/uncommon toxicity
- Drug-drug interactions
- Applicable for evaluating drug selection
- Oncologists “vote with their feet”
- Reasonable approach to evaluate outcomes
|
RWD is typically analyzed by epidemiologists. Pharmacoepidemiology is a specialized field of epidemiology, and pharmacoepidemiologists study the utilization and outcomes of drugs in the real world and provide estimates of the beneficial or harmful effects in a population. There are multiple study designs that can be considered in the evaluation of RWD. Currently, the FDA predominately uses descriptive statistics to analyze RWD. This is evident in the prescribing information of drugs, which generally includes a heading similar to “post-marketing experience” and a disclaimer about an uncertain denominator and the voluntarily reporting of data.
Academia generally use more advanced epidemiology study designs that are defined as cohort studies or case-control studies (Figure 2). When moving beyond descriptive studies, the easiest way to classify the analysis is to begin by considering the point at which a patient is selected to be in the analysis. If the study collects patients who have already had an outcome and then evaluates exposures prior to that outcome, it is classified as a case-control study. If, instead, patients are included when they have an exposure and are then followed to see if they develop an outcome of interest, the study is a cohort design. A simple way to think about a cohort study design is as a prospective trial with existing data. For example, a metastatic breast cancer patient receives hormone therapy with or without a cyclin-dependent kinase (CDK) 4/6 inhibitor: she would be followed to a pre-determined endpoint such as duration of therapy or progression as a marker of efficacy. This is the most common type of pharmacoepidemiology study design with RWD.
Figure 2. Study Designs to Evaluate Real-World Data
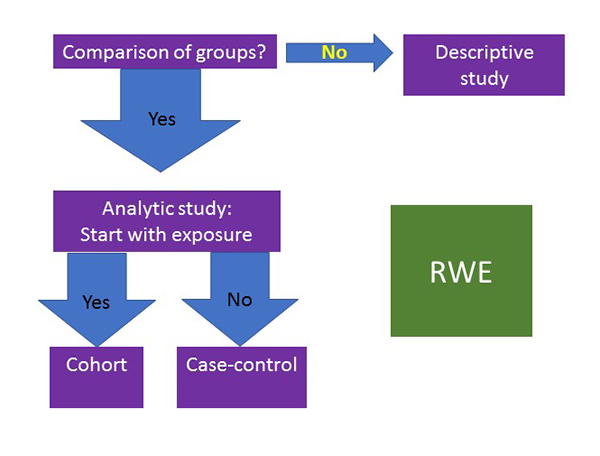 |
| RWE, real-world evidence. |
Potential applications of study designs to augment FDA approvals
Several areas of development and guidance are critical when considering how the 21st Century Cures Act could utilize RWD to support FDA submissions with the goal of accelerating development and advances to patients.5 For approved drugs, it is easy to imagine that analyses of real-world practices could lead to an expansion of the approved uses of a drug. This would be particularly helpful in the setting of uncommon or rare diseases. Future CDER guidance should provide some indication as to the threshold needed for such an approval. Namely, could RWD be strong enough to expand an indication, which could minimize the amount of research currently required for approval, or would a new indication require a new clinical trial? Another potential use of RWE is as a control population for comparison. This type of approach is a blend of RWE and a clinical trial. This could especially benefit pediatric research—a field in which physicians are already using drugs without clinical trial data.
Pharmaceutical companies may also seek to expand prescribing information for an approved drug, which would require the real-world study of populations excluded from the original clinical trial. In the realm of oncology, that commonly includes patients with brain metastasis and/or significant organ dysfunction. Since many of these patients still desire treatment, oncologists will commonly extrapolate clinical trial data to develop a treatment plan. Study of this real-world use could establish efficacy and safety and, in the situation of organ dysfunction, dosing.
Assessing the validity of trials
Although the details of how the FDA plans to use RWE are not entirely known, it is clear that change is coming: the study designs used in the future will be stronger than the descriptive statistics currently used. Most clinicians have become knowledgeable consumers of clinical trial study designs and methodology; as such, more complex epidemiology study designs will pose a challenge: it may be difficult for clinicians to understand the strengths and shortcomings of the studies’ conclusions. The benefits of the FDA incorporating epidemiology study designs into its review and approval processes will likely require an investment from pharmaceutical companies. Although there are potential biases due to financial interests in the outcomes, there is benefit that the research will be submitted to and endorsed by the FDA. Because research is generally very expensive, the only business model that supports approval comes from the pharmaceutical industry. Generally, the research they support and perform is approved by the FDA. Rather than planning and performing millions of dollars of research only to discover the FDA will not accept the study design or endpoints, pharmaceutical companies generally consult with the FDA before embarking on a trial. With FDA oversight upfront, as well as its review of the final results, pharmaceutical companies are accountable for performing high-quality research. Statisticians are an integral part of the industry’s research team and the FDA’s review team. Consequently, clinicians should feel confident in a study’s results if it is industry funded and FDA reviewed/approved.
For academia-initiated studies of RWD that utilize complex study methodologies, signs of quality include (1) having a statistician involved to such an extent that he or she is an author of the study and (2) publication in a high-quality journal, which would likely conduct a statistician review of its own. These factors are important, but equally important are the impressions of clinicians on the basis of study questions, controls, and endpoints. Clinicians, better than statisticians, understand the disease, pathology, pharmacology, and real-world issues related to treatment: they should make sure the questions being addressed are important and that the endpoints selected make sense. A tool is available accompanying this monograph that can be used as a quick guide to evaluate the quality of an epidemiology study.
RWE IN BREAST CANCER
Breast cancer is a common disease that is estimated to be diagnosed in more than 250,000 people in the U.S. in 2018. The majority of these patients will have localized disease and approximately 6% will have metastatic disease at initial diagnosis. For those with local disease, surgical resection (lumpectomy or mastectomy) will lead to complete remission; however, adjuvant therapy is still routinely administered due to the undetected micrometastases that historically return.9,10 Even with adjuvant therapy, some patients will have a recurrence of breast cancer. Recurrence-free survival at 10 years for stages I, II, and III disease was 80.5%, according to results of landmark trials that treated 2838 patients with adjuvant or neoadjuvant therapy. Many of the recurrences are metastatic, which adds to the total number of patients with metastatic breast cancer.11
Analysis of the tumor and assessment of mitogenic drivers are required for selection of treatment and determination of prognosis. The choice of therapy for all stages of breast cancer is based on the tumor expression (overexpression) of hormone receptors (HRs), including estrogen receptors (ERs) and progesterone receptors (PRs), and human epidermal growth factor receptor-2 (HER2). These characteristics allow patients to be categorized into 1 of 4 groups that guide therapy. One group is known as “triple negative,” which, as the name implies, includes tumors with minimal or no , PR, and HER2 expression. The only treatment option for patients with this subset of breast cancer is chemotherapy. A second group includes HR-negative (HR–) (i.e., both ER-negative [ER–] and PR-negative [PR–]) and HER2-positive (HER2+) tumors. Patients with this type of breast cancer should be treated with chemotherapy plus a HER2-directed therapy (e.g., trastuzumab). A third group is termed “triple positive,” indicating that the tumor expresses both HRs (i.e., ER-positive [ER+] and PR-positive [PR+]) and HER2. Treatment for this type of cancer should include hormone therapy, as well as anti-HER2 therapy and chemotherapy. The last subset of breast cancer is HR-positive (HR+), HER2-negative (HER2–) disease. Patients with this type of cancer should be treated with hormone therapy, with or without chemotherapy. HR+/HER2– tumors dominate breast cancer biology, with an estimated 3 out of 4 tumors fitting in this category.12,13
Disease stage affects the goals of breast cancer therapy and, consequently, the treatment approach.14 The goal of treatment of localized and regional disease is cure. Therefore, patients with this type of disease generally receive aggressive therapy to kill any subclinical micrometastatic disease. Metastatic disease cannot be cured, so the goal of therapy for this type of cancer is the prolongation of the number of quality days, months, or years that a person will live. Historically, this translates into a treatment approach that includes single-agent therapies administered sequentially. Because hormone therapy is less toxic than chemotherapy (and HR+ tumors are the most common type of tumor), it is used as the first-line choice for most patients. The exception to this rule is patients who have visceral organ disease or rapidly growing tumors: these patients should receive chemotherapy. The rationale for this approach is based on the fact that tumor response is needed to improve symptoms and quality of life. The time to response is much faster for chemotherapy than for hormone therapy. Patients who respond to first-line hormone therapy but then experience disease progression will typically receive second-line hormone therapy with a different class of agents. This sequential use of hormone therapy continues until patients fail to respond to 2 consecutive treatments. This historical approach is now evolving due to other targeted therapies that work synergistically with hormone therapy and can be less toxic than chemotherapy and hormone therapy. This module’s discussion of RWE will focus on the metastatic breast cancer population and evaluate cases in which an oncologist may prescribe a CDK 4/6 inhibitor outside of the FDA-approved labeling.
CURRENT TREATMENT RECOMMENDATIONS FOR METASTATIC HR+/HER2– BREAST CANCER
Women with stage IV and recurrent HR+/HER2– breast cancer are categorized as premenopausal or postmenopausal, as drugs and treatment options vary with these designations. The primary reason for this is the high incidence of premenopausal women who produce estrogen despite the administration of an aromatase inhibitor. Additionally, prior adjuvant therapy with hormone therapy within the last year can affect drug selection. Essentially, National Comprehensive Cancer Network (NCCN) guidelines suggest that if a patient is resistant to the last line of therapy, then a different class of agents should be used. Patients with visceral organ involvement may require chemotherapy to generate a faster response. Other treatment options are outlined in Figure 3.12
Figure 3. Treatment Selection for Newly Diagnosed Stage IV or Recurrent HR+/HER2– Breast Cancer12
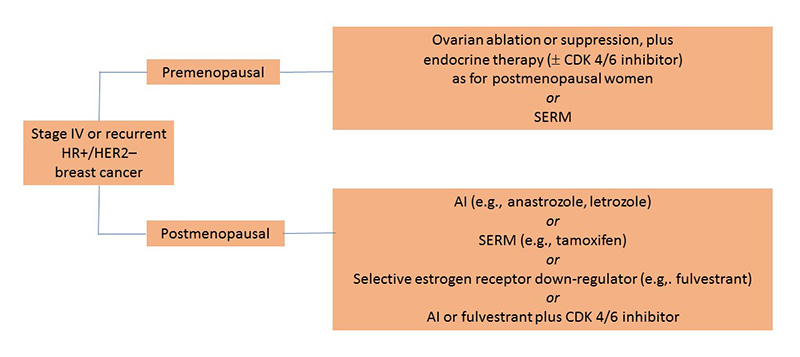 |
| AI, aromatase inhibitor; CDK 4/6, cyclin-dependent kinase; HER2–, human epidermal growth factor negative; HR+, hormone receptor positive; SERM, selective estrogen receptor modulator. |
CDK 4/6 inhibitors
In February 2015, palbociclib became the first CDK 4/6 inhibitor approved in its class, and it paved the way for ribociclib and abemaciclib. All 3 agents are approved for the treatment of metastatic or recurrent HR+/HER2– breast cancer. They are approved for use with an aromatase inhibitor as first-line therapy or with fulvestrant as second-line therapy. (Ribociclib is also approved for use with fulvestrant as first-line therapy.) These agents are detailed in Table 315-17 and in the clinical tool provided with this module. The pharmacology is the same for all 3 agents, but the toxicity profiles and recommended monitoring parameters differ.
CDK 4/6 and cell division
During the development of CDK 4/6 inhibitors, it was discovered that they work best in luminal A tumor cells, which are HR+ and HER2–. As part of the drug development, researchers investigated how tumors become resistant to hormone therapy and how CDK 4/6 inhibition could augment or resensitize cells to hormone therapy. In short, cell signaling follows the results of positive and negative signals as follows: HR+ breast cancer cells use estrogen as a mitogenic factor; essentially, estrogen binds to the ER in the cytoplasm, then the complex moves to the nucleus to stimulate cyclin D expression; cyclin D binds and activates CDK 4 and CDK 6, which, in turn, phosphorylates retinoblastoma protein (RB); the phosphorylated RB can no longer bind to E2F and the unbound E2F allows this transcription factor to stimulate the cell past the restriction point in G1 of the cell cycle and commits it to cell division. Inhibiting estrogen, therefore, is an effective treatment for these types of tumor cells; however, resistance occurs over time and generally results from the tumor cell finding a different way to activate the cyclin D-CDK 4/6 complex to carry the signal. The combined targeting of estrogen and CDK 4/6 has been shown to prolong the time for the cancer cell to develop resistance or it can be used after resistance to hormone therapy is established.18-21
| Table 3. Characteristics of CDK 4/6 Inhibitors15-17 |
| |
Abemaciclib |
Palbociclib |
Ribociclib |
Approved indication |
HR+, HER2–, Adv, or Met breast cancer |
HR+, HER2–, Adv, or Met breast cancer |
HR+, HER2–, Adv, or Met breast cancer |
Regimen(s) |
150 mg PO BID with AI 1st line or fulvestrant 2nd line
or
200 mg PO BID as monotherapy |
125 mg PO daily x 21 days followed by 1 week off
with AI 1st line
or with fulvestrant 2nd line |
600 mg PO daily x 21 days followed by 1 week off
with AI or fulvestrant 1st line |
Administration |
Take regardless of food |
Take with food |
Take regardless of food |
Drug-food interaction |
Increases AUC by 9% |
Increases AUC by 21% and decreases variability
Avoid taking w/ grapefruit juice |
Food has no effect |
Drug-drug interaction |
CYP3A4 substrate: avoid strong inhibitors/inducers |
CYP3A4 substrate: avoid strong inhibitors/inducers |
CYP3A4 substrate: avoid strong inhibitors/inducers |
CBC with differential monitoring |
Baseline, every 2 weeks for 2 months, then monthly for 2 months |
Baseline, every 2 weeks for 2 months, then monthly for 4 months, then every 3 months |
Baseline, every 2 weeks for 2 months, then monthly for 4 months |
Liver function tests (AST, ALT, bilirubin) monitoring |
Baseline, every 2 weeks for 2 months, then monthly for 2 months |
NA |
Baseline, every 2 weeks for 2 months, then monthly for 4 months |
Serum electrolytes (K+, Ca2+, Mg2+, Phos) monitoring |
NA |
NA |
Baseline, monthly for 6 months |
ECG monitoring |
NA |
NA |
Baseline, day 14 of cycle 1, day 1 of cycle 2 |
| Adv, advanced; AI, aromatase inhibitor; ALT, alanine aminotransferase; AST, aspartate aminotransferase; AUC, area under the curve; BID, twice daily; CBC, complete blood count; CDK, cyclin-dependent kinase; CYP, cytochrome P450; ECG, electrocardiogram; HER2–, human epidermal growth factor negative; HR+, hormone receptor positive; Met, metastatic; NA, not applicable; PO, by mouth. |
