
Expired activity
Please go to the PowerPak
homepage and select a course.
Exploring Emerging Strategies in the Management of Anemia in Chronic Kidney Disease
A Closer Look at the Burden of CKD-Associated Anemia and Rationale for HIF-PH Inhibition as a Therapeutic Strategy
Dr. Wish: The name of the program is “Exploring Emerging Strategies in the Management of Anemia in Chronic Kidney Disease.” And today’s panelists are myself, Anil Agarwal from Ohio State, and Dr. Thomas Dowling. Our first member of the panel, Dr. Anil Agarwal, will give us a closer look at the burden of CKD-associated anemia and the rationale for HIF prolyl-hydroxylase inhibition as a therapeutic strategy. Anil?
Slide 1
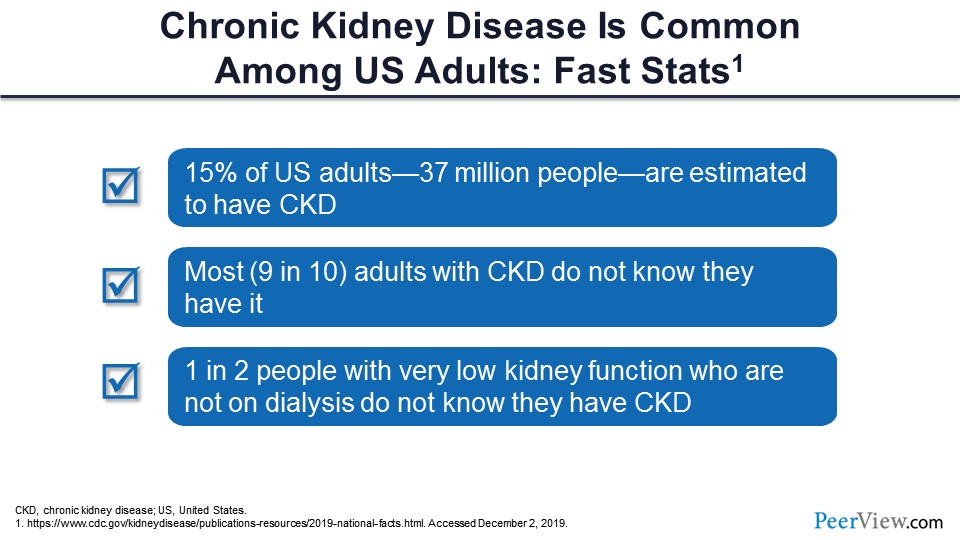
Dr. Agarwal: What we are going to do today is discuss the scope of anemia and how it is important, to learn the strategies to treat it well—not just the old paradigm. What is the new paradigm that is coming? So this is just to refresh you. I know you all know about this; there are data from the CDC. CKD is actually very common. About 15% of US adults—that comes to about 37 million people—are estimated to have CKD. This may be a slight over- or underestimation. What is more striking is that 90% of these people who have CKD are actually not aware that they have CKD. And even more striking is that people who have a very advanced degree of CKD, sometimes they come in, and they don’t know. About 50% of those people do not know that they have that degree of CKD. It’s totally not unusual for me to see some patients who come with CKD stage 4, and when I tell them, “You have kidney disease,” they have no clue that they have kidney disease.
Slide 2
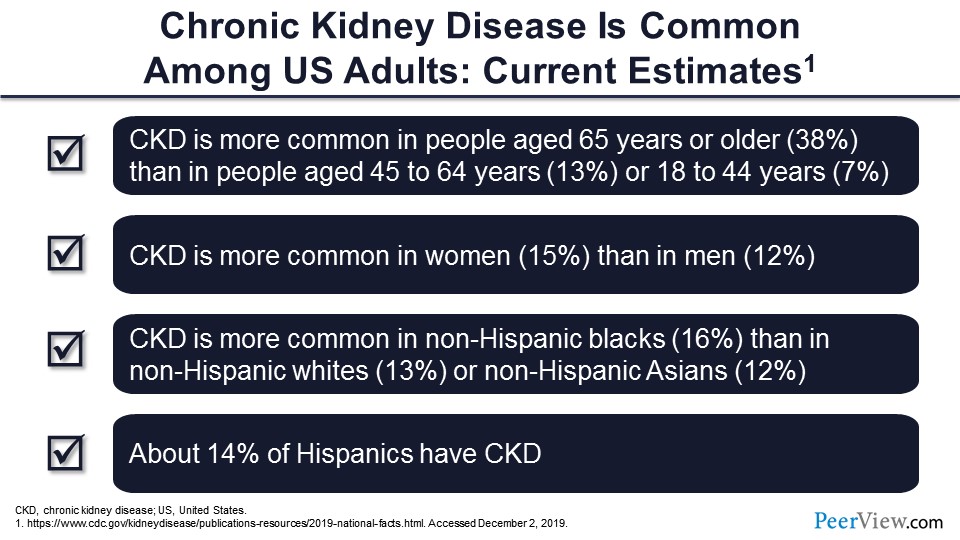
CKD is more common as you get older. After 40 years of age, you start losing kidney function—1% almost every year—so it’s more common to have it in older age. Contrary to popular belief, it is more common in the ladies. And it is, of course, more common in non-Hispanic blacks than non-Hispanic whites and Asians. And about 14% of Hispanics also have CKD.
Slide 3

So what about anemia? We all know that as the kidney function goes down, you start producing less erythropoietin, and you become anemic. Anemia is an important complication that is very, very common and that affects the life of our patients with CKD. However, the bright side is that this is something we can treat. Of course, we don’t know apparently how to treat it very well; but this is something that is treatable, and that’s why we are here today.
When anemia is present, it is not without symptoms. You feel fatigued, you have bad cognition, you have weakness, and sometimes shortness of breath and cardiac complications. Also, how many people have anemia is defined by how you define anemia. So, if you say anybody below the hemoglobin of 13 g/dL has anemia, you will have more people having anemia. If you say 12 g/dL, you will have a lower number of people having anemia. There are different definitions beyond the scope of today’s discussion. The World Health Organization has a different one. KDOQI has different one, and we’ll go a little bit over that.
Slide 4
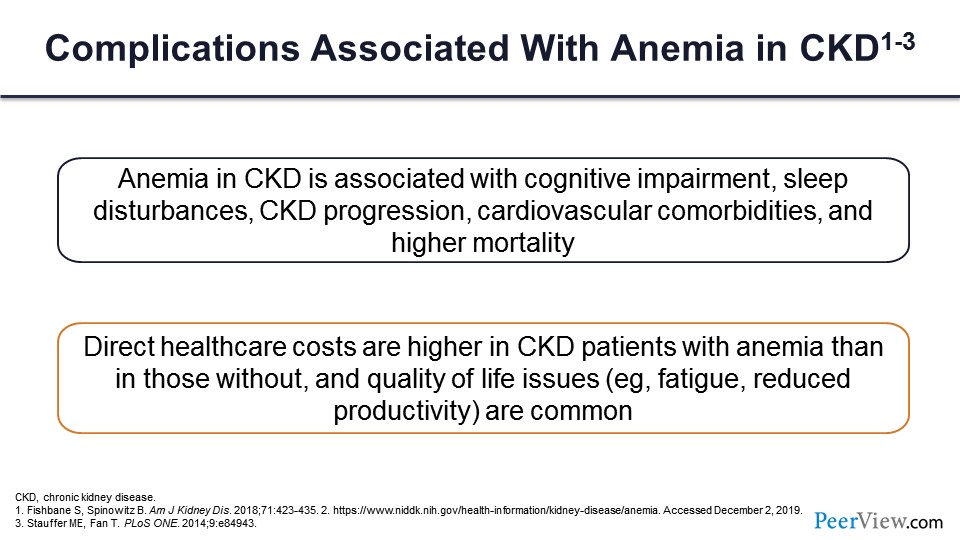
When you have anemia, it is not without complications. As I was mentioning, the effects are serious. How much oxygen is getting to the brain is important, so cognitive impairment, sleep disturbances. Sometimes people have attributed CKD progression to higher degrees of anemia, cardiovascular comorbidities, and of course, higher mortalities are associated with anemia.
Not only is there a human cost, but there is also a financial cost. I’m sure you all are very aware of how expensive the ESAs are, and iron is not cheap either. So, the direct healthcare costs are higher for patients who have anemia with CKD than for those who do not. And people who have anemia are fatigued and not that productive, so you have further loss of productivity associated with that.
Slide 5
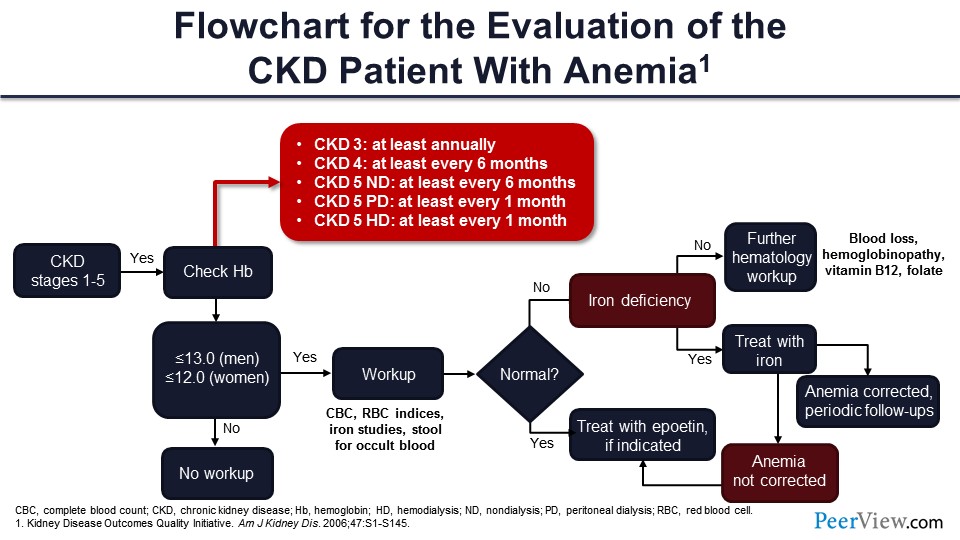
You remember the KDOQI guidelines—the KDOQI came from the Kidney Disease Outcomes Quality Initiative back in 2006. The National Kidney Foundation published this table. So first of all, you check hemoglobin—and this is anybody from CKD stage 1 to 5. If you have hemoglobin less than 13 g/dL in men and less than 12 g/dL in women, now you have anemia, and you need to start a workup.
Of course, you start with the CBC, the RBC indices, the iron, and the stools. You want to make sure the patient is not bleeding. And if you have iron deficiency, you can treat it. There’s nothing else to be done. But if you don’t have iron deficiency, you want to make sure the patient does not have cancer, does not have GI bleed, so B12, folate—you have to check all those.
Once you have done all that and everything else is normal, the inference is this patient has CKD, they have anemia. There is no other cause. It must be erythropoietin deficiency, and you can start treatment with erythropoietin.
Slide 6
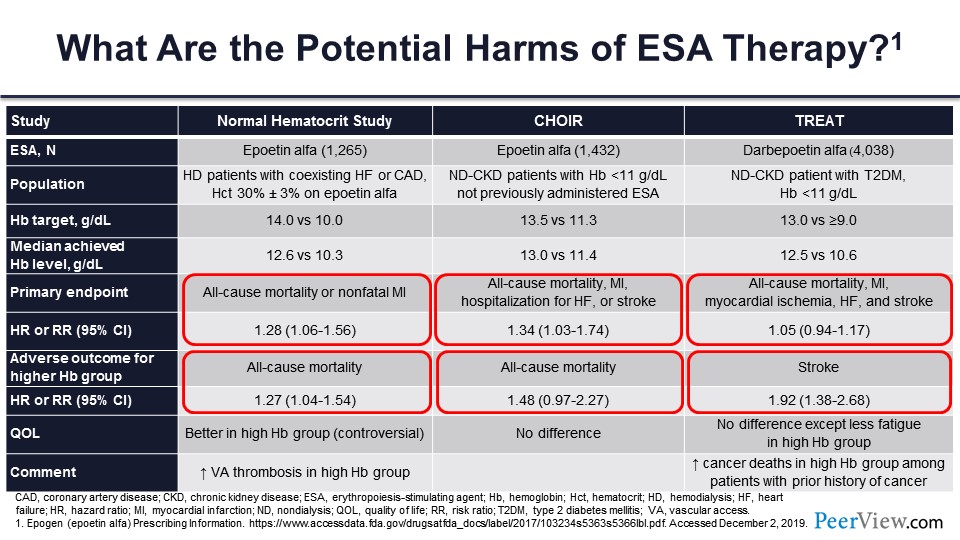
What’s the problem with that? It’s simple: You have less erythropoietin, you give erythropoietin, you get better hemoglobin, and you are done. But there is potential harm with ESA therapy. There have been a number of randomized, controlled trials—I’m not just talking about observational trials.
Some of you who are getting toward my age will remember the Normal Hematocrit Study. This tested epoetin-α in patients with heart failure. CHOIR tested epoetin-α in nondialysis-dependent patients with CKD. And TREAT, tested darbepoetin in nondialysis-dependent patients with CKD who had diabetes also. And they all targeted a higher level of hemoglobin in one arm and lower level of hemoglobin in the other arm. You can see they are a little different, but all are a comparison with target hemoglobin.
All these studies found that the composite outcomes of all-cause mortality and cardiovascular events were generally much higher in the Normal Hematocrit Study and CHOIR. In TREAT, there was not so much of a difference, but the all-cause mortality both in Normal Hematocrit Study and CHOIR was 27% higher and 48% higher, respectively.
Quality of life (QOL), that’s kind of a controversial subject; how to measure QOL is not the easiest thing. It was better in the high hemoglobin group, no difference in CHOIR, and there was no difference—although there was less fatigue—in TREAT. There were other complications: more vascular access thrombosis in Normal Hematocrit Study and increased number of cancer deaths in those people who had a history of cancer in patients who were enrolled in TREAT.
Slide 7
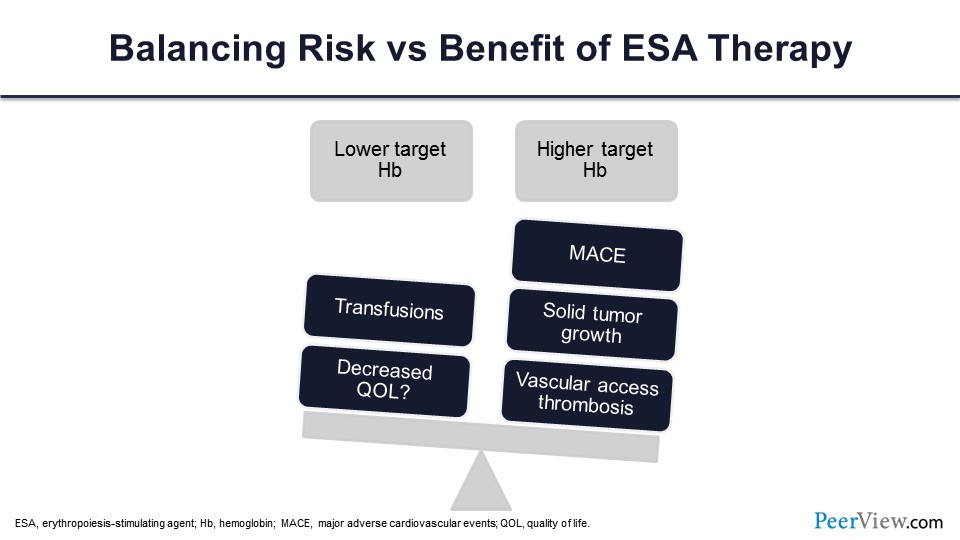
You’ve got a very delicate balance when you are using ESA. You have to balance the lower target hemoglobin with the higher target hemoglobin. When you do lower target hemoglobin, you are risking transfusions and decreasing QOL. On the other hand, if you shoot for the higher target, you have major adverse cardiovascular events (MACE), cancers, and vascular access thrombosis.
Slide 8
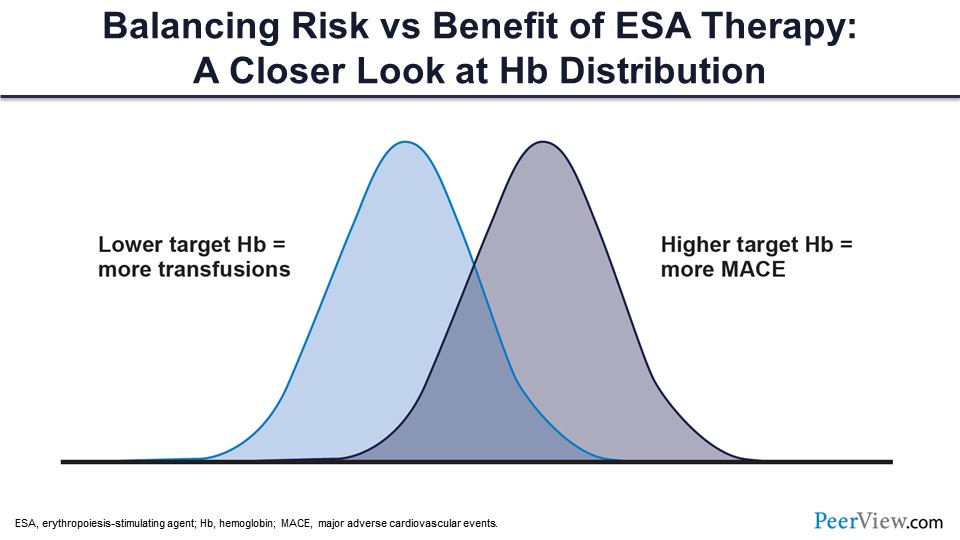
So this is something that you have to keep in mind, and you have to strike this sweet spot somehow where you don’t run into transfusions and don’t cause more cardiovascular events.
Slide 9

Considering the results of all those studies that showed higher target hemoglobins are not good, the FDA issued an advisory back in 2011 that made an ESA label change. After that, they said, “You cannot use it to keep hemoglobin at 12 g/dL,” which was usual for people before that. They said, “Okay, you should probably either interrupt or stop the treatment at 11g/dL.”
The use of ESA dosages actually got down to about 40% of the previous dosages. Although it was associated with stroke, venous thromboembolism, and heart failure, incidences decreased—so something good, and something bad.
The mean hemoglobin levels decreased from 11.5 to 10.8 g/dL. Transfusions increased. And at the same time, it is very evident from this that you are trading off some benefits with some harms when you give higher dosages of ESA to achieve higher targets. Is that acceptable? And this didn’t even necessarily consider the QOL.
Slide 10
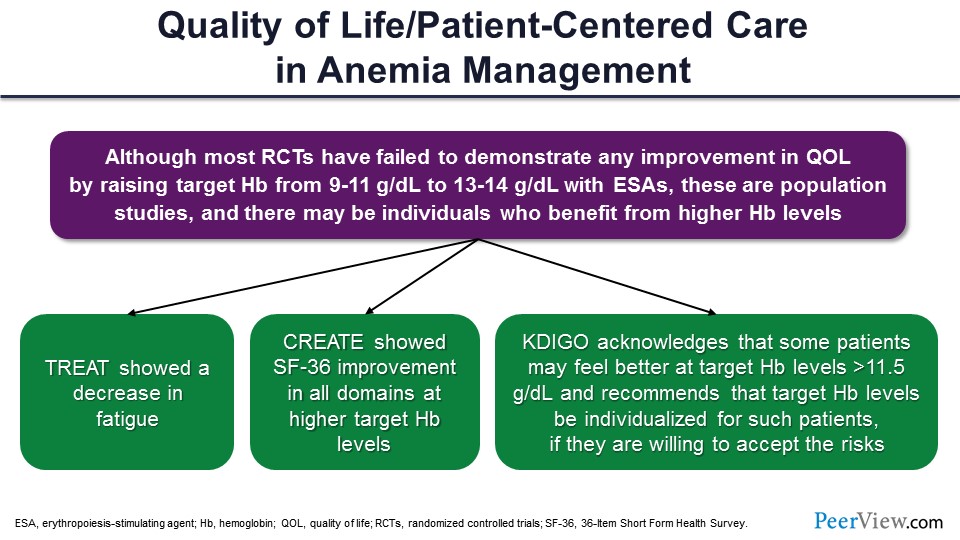
We know that most of the randomized controlled trials (RCTs) did not show that there was any change in QOL with higher targets; but at the same time, that’s not personalized care. You cannot treat everyone the same. There may be some people who feel better at higher levels of hemoglobin, and they will say, “My hemoglobin is down, and I’m not feeling well.” For example, TREAT showed less fatigue when the hemoglobin was better. CREATE showed improvement. And the KDIGO guidelines also suggested that some people may require hemoglobin greater than 11.5 g/dL, and you have to individualize considering that there are risks.
Slide 11

So what are the proposed mechanisms? Nobody knows what exactly causes those bad outcomes, but because we randomized people to either a higher target or a lower target, we have to infer that the higher targets were probably the cause. But at the same time, most of the studies have shown an association of higher ESA dosages with occurrence of adverse events in secondary analysis.
At the same time, you have to keep in mind that people who had high hemoglobin without using ESA actually had the best outcomes; so by itself, high hemoglobin is not bad. By giving high doses of ESA to drive hemoglobin to a higher target, that’s where the problem comes in. This may be related to a high number of comorbidities, the inflammation that comes with CKD that might be causing ESA resistance, and pharmacologic use of erythropoietin that might be causing all those problems.
Slide 12

What may be the other mechanisms? You raise hemoglobin, you increase the viscosity, and you cause margination of platelets. There might be more thrombotic events because of that, and there may be decreased peripheral vasodilatation, maybe a slight increase in the blood pressure, endothelial dysfunction. And at that point, because of the endothelium and angiotensin release, you might have a defective function of the smooth muscles, which might be causing all these adverse effects. And plus, when you give ESA, and if you are iron deficient, there is a platelet response, so you get thrombocytosis. And that also can result in thrombotic events.
Slide 13

Also, we must think in terms of how high the dosages we’re using are. If you take a normal person with normal hemoglobin, the erythropoietin level is very minuscule; it’s 4 to 24 mU/mL. Whereas, if you give 30 U/kg of erythropoietin intravenously, what you will find is that the erythropoietin level goes to 600 mU/mL. That’s something. That’s a very high level. And when you have high levels of erythropoietin, it might be affecting other receptors than just the erythropoiesis receptor, so you might be affecting receptors in heart, brain, CNS, and vasculature, which might be cytoprotective but might also be trophic, which might cause growth. So for example, if you keep stimulating the receptors in the myocardium, you might get more left ventricular hypertrophy and less remodeling. And you know, left ventricular hypertrophy by itself is a risk factor for mortality.
Slide 14
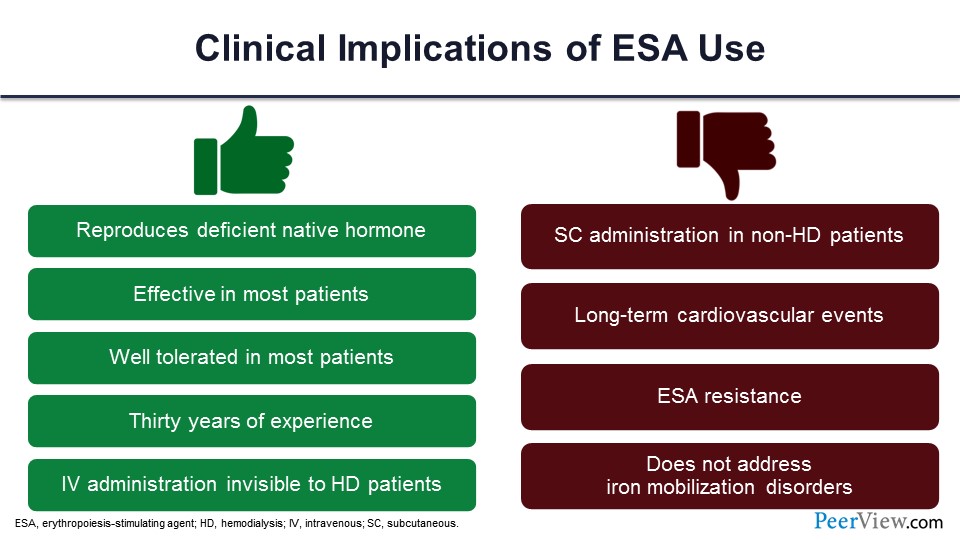
To summarize the ESA effects, there are clinical implications. There are some good things. You are replacing the hormone. It is effective. It’s well tolerated. You can give it intravenously to patients on dialysis, and they don’t feel it. It is a time-tested drug. We have been using it for a long time. At the same time, if the patients are not on dialysis, you have to give it subcutaneously, and we have seen long-term cardiovascular events. We have seen those adverse cardiovascular events. ESA resistance can occur. And it really doesn’t do anything to the iron or the inflammation, so there are pros and cons of that.
Slide 15

The second model of treatment of anemia in CKD is to give them iron. Well, they are iron deficient, they are erythropoietin deficient, you gave ESAs, and now you need to give them iron. In nondialysis-dependent patients with CKD, iron deficiency is very, very common. It’s recommended they are given a trial of either oral or IV iron. IV iron works better; it gets directly into the system, and you don’t have the GI-related adverse events. IV iron, on the other hand, does have some adverse events. You can have anaphylactic reaction, for example. There could be allergic reactions.
In hemodialysis, we know that we give a lot of iron; about 80% of patients end up getting one or the other form of IV iron because there are ongoing losses in the dialysis circuit. And these patients have platelet dysfunction, so they bleed also more commonly. They have inflammation; and because of inflammation, their ESA does not work well. So you have to give higher doses of ESA, but that causes more problems with the iron that I am coming to.
Slide 16

So, just a word about IV iron. You know that there are benefits, which I already went over. There are also risks for IV iron. There could be more inflammation and oxidative stress. And some people have attributed cardiovascular events to iron as well, and higher mortality because of that. So, we just don’t know.
Slide 17
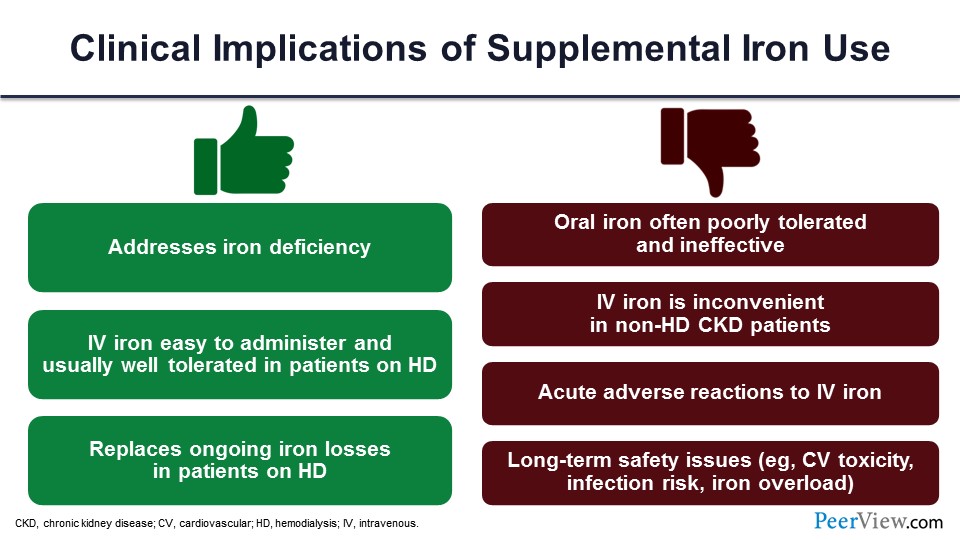
There are pros and cons of iron use, as well. Yes, you are replacing the iron, which was deficient. It’s easy to administer IV iron. And if you give iron, you reduce the ESA dosages; we know that. But oral iron is not very well tolerated. IV iron has its own adverse events. There may be long-term safety issues with the iron. There could be iron overload, if you give too much. So, all those things are also on the table. As you can see, we don’t really have a good paradigm for treating anemia in CKD just using ESA and iron.
Slide 18
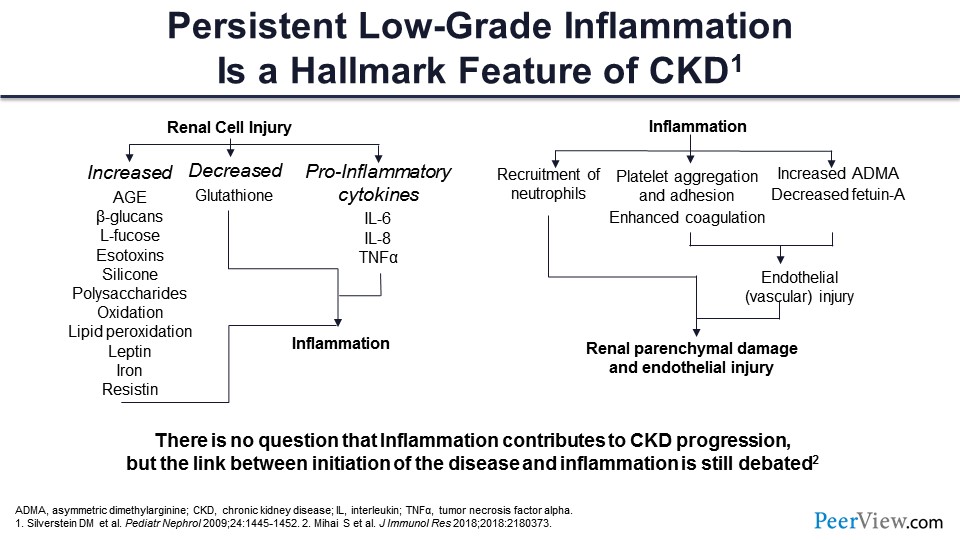
What about the inflammation? Anybody who went to med school or school before 2000 wouldn’t really know this history, and we were not taught this in med school because hepcidin was discovered in about 2000. What characterizes CKD is inflammation. There are two ways it works. When you get kidney injury and the kidney function goes down, you get accumulation of advanced glycosylation and products, as well as multiple other molecules that lead to oxidation and lipid peroxidation and cause membrane damage. There is decreased glutathione. There are pro-inflammatory cytokines, so there is big-time inflammation going on in patients with CKD.
On the other hand, if you have inflammation that recruits neutrophils, that can cause kidney damage. Inflammation leads to platelet aggregation and adhesion, probably resulting in those thrombotic risks. It also causes dysfunction of the endothelium. There is increased asymmetric dimethylarginine, and fetuin-A is decreased, which also causes endothelial damage. Now your inflammation feeds into the kidney disease, and the kidney disease feeds into the inflammation, and you have this perpetual cycle of kidney damage and inflammation that goes on. We don’t know if inflammation by itself causes kidney damage, but we definitely know that with inflammation kidney disease gets worse.
Slide 19
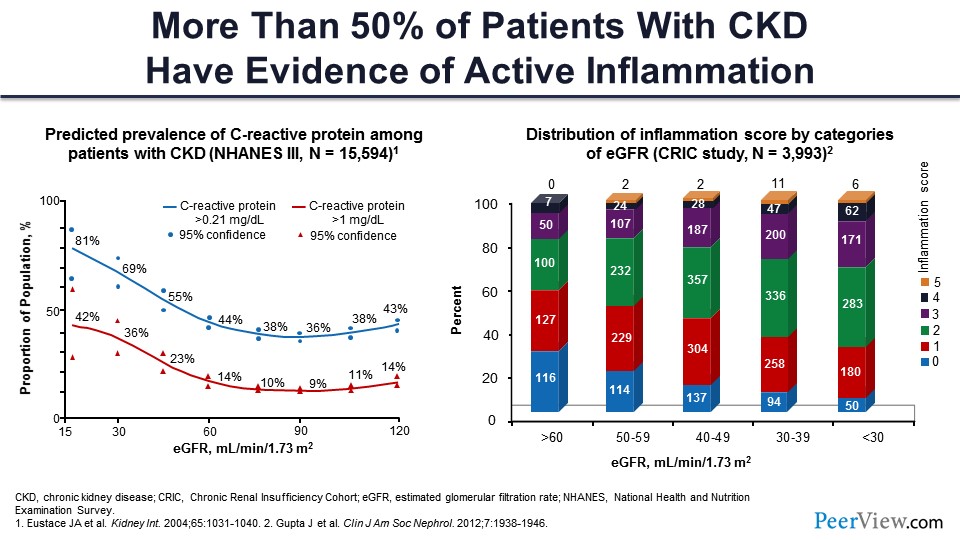
People have actually looked for the evidence of inflammation. If you look at C-reactive protein levels as the glomerular filtration rate (GFR) goes down, you can see that the C-reactive protein CRP level goes high. And about 80% of people have about 1 mg/dL of CRP as the serum creatinine goes up and GFR goes down. There is another study called the CRIC study—the Chronic Renal Insufficiency Cohort—that is multicenter study with 4,000 or 5,000 patients. They have been following these patients for multiple, different parameters. They looked at five different markers of inflammation in their cohort, and what they found is that as the GFR goes down from 60 down to less than 30 mL/min/1.73m2, the number of people with five parameters or more were higher, indicating that there was a higher degree of inflammation. They called it an inflammation score. So, the inflammation score was higher in patients with worse kidney function.
Slide 20
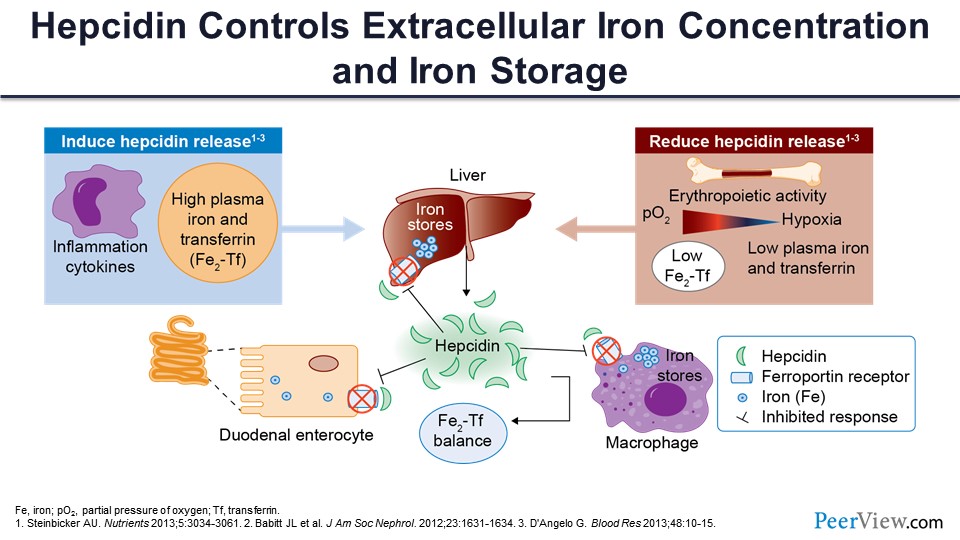
So, what happens with that? When you have inflammation, there is secretion of hepcidin by the hepatocytes. It’s a 25-amino-acid peptide; it is part of the innate immunity, and it was meant to prevent bacterial infection. So, it was usually secreted in the response to any infection or inflammation. So what happens now? Hepcidin says, “Okay, the bacteria like iron; we don’t want any iron in the circulation. Let’s lock it down.” There is something called ferroportin, a very important protein that takes iron outside the cell. So, when hepcidin is released, it blocks the ferroportin, and the iron stays in the liver. It also blocks the duodenal enterocyte ferroportin, which reduces the absorption of iron, so you get less iron. It also locks up the iron within the macrophage and the spleen and reticuloendothelial system, and now you cannot get any iron. Now you have somebody who is anemic but inflamed, and they cannot synthesize more hemoglobin because the free iron is not there. Even though there is iron in the store, it’s just not been released. And in any state where you have inflammation or high plasma iron, there is more hepcidin released. And if you have a situation where you want more erythropoiesis and you are iron deficient, hepcidin secretion actually goes down. So hepcidin is the key regulator. It really locks up the iron within the cells if there is inflammation.
Slide 21
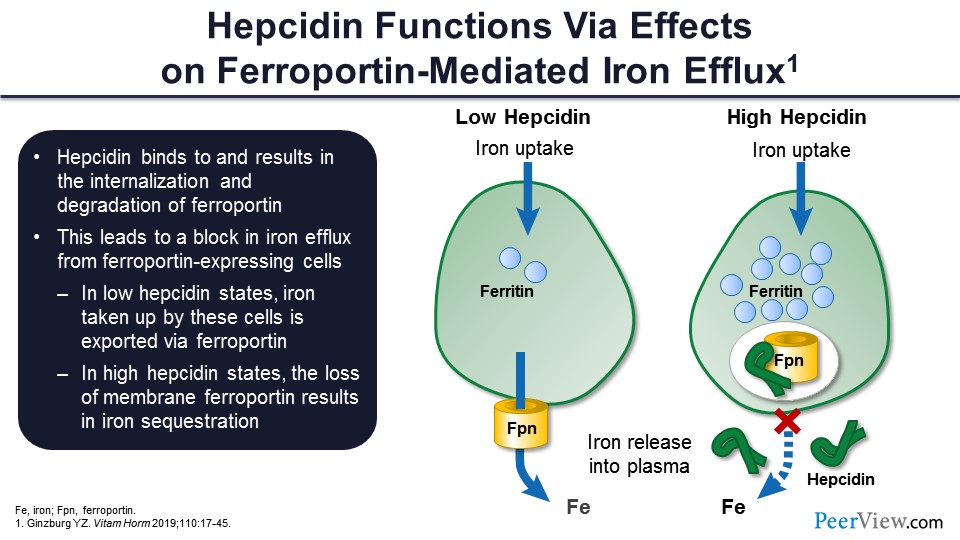
And, as I was mentioning to you, it works through ferroportin. When there is low hepcidin, the ferroportin takes the iron out of the cell. It becomes available for erythropoiesis. When there is high hepcidin, it sort of blocks down the ferroportin, and ferroportin is then degraded. You get a lot of iron inside in the form of ferritin, but you don’t have the iron attached to transferrin, and you don’t have iron that goes for erythropoiesis.
Slide 22
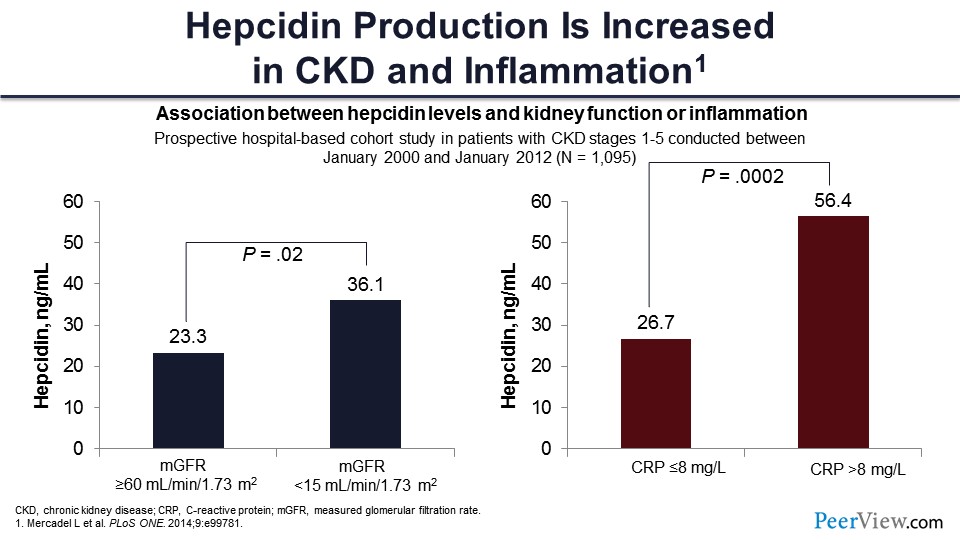
We know that there is inflammation, and hepcidin causes problems with iron delivery. So people have looked at the level of hepcidin in patients with kidney disease. You can see these are people with GFR greater than 60 mL/min/1.73m2, and this is the hepcidin level of 23 mL/min/1.73m2 here. When the patients have less than 15 mL of GFR, the level is higher in those patients.
Similarly, if you have patients with lower CRP, less than 8—8 is actually high—but still, if they have less than 8 ng/mL, this is the hepcidin level. But if you have CRP greater than 8 ng/mL, the level is more than double. So here, the common theme is you get inflammation, you get more hepcidin, you lock up more iron, and you’re not able to use iron for erythropoiesis.
Slide 23
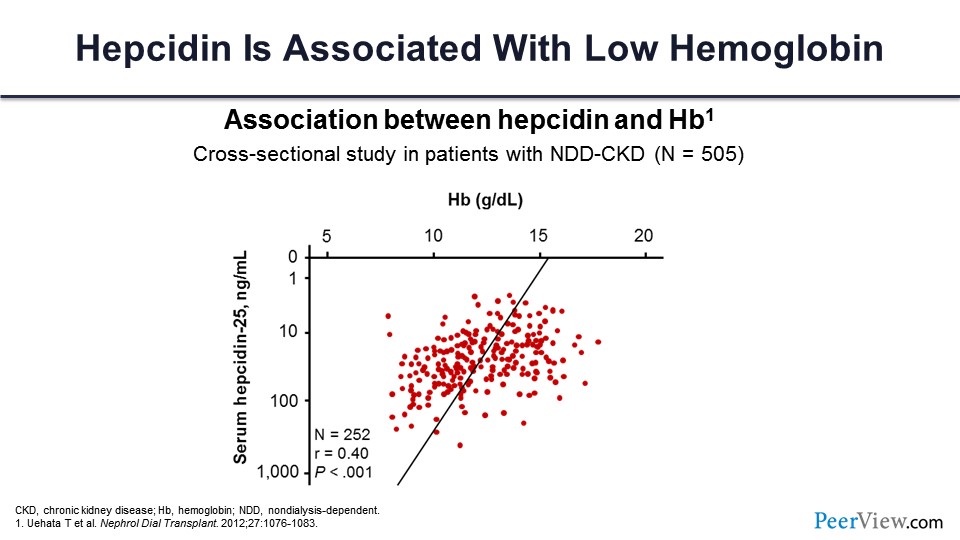
And the consequence is as the hepcidin level goes up—this is the level going up and this is the hemoglobin going down with that. So, they correlate very well together.
Slide 24

There is a difference between the absolute deficiency of iron and a functional deficiency of iron. And you don’t have to look anywhere else—kidney disease is the perfect model for every type of iron deficiency. Patients are not eating, they don’t feel good, and they are iron deficient on that basis. Then we give them ESA, and ESA actually starts using up the iron, and they become deficient on that. And then they have inflammation, they have iron, and their iron is not being used because it’s not available to the erythron.
So this is a pictorial representation of a normal person. Storage iron is adequate, transport iron is adequate, and there is iron for erythropoiesis. This is the normal condition. This is how everybody should be. But if you have absolute iron deficiency, you have depleted the storage iron, there may be less transport iron as well, and there is less iron available to erythron. In functional iron deficiency, the difference is that you have a lot of ferritin available. The storage iron is complete—it’s there—but you don’t have much of the transport iron, and there is less iron available to the erythron, even though you have a good store of iron.
Slide 25
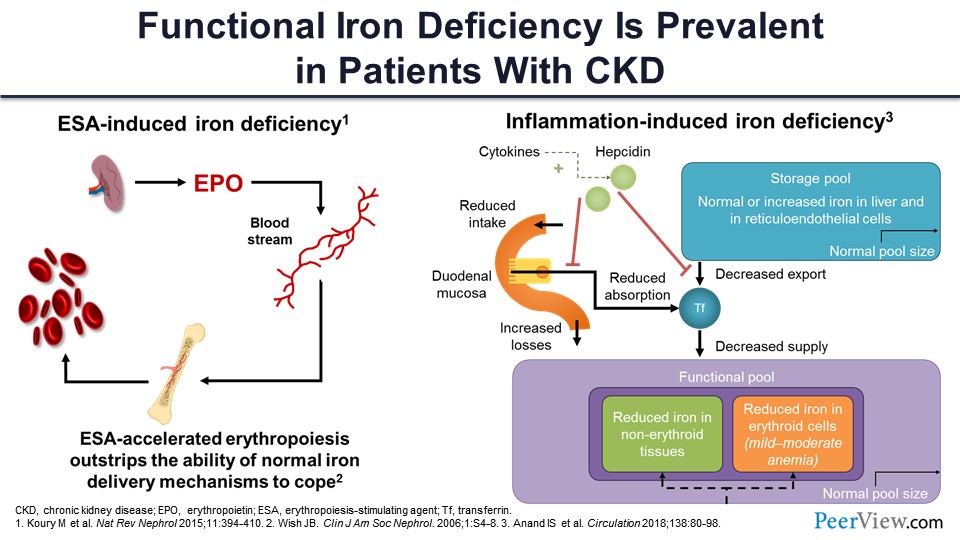
I was telling you about the iron deficiency again. So this is the ESA-induced iron deficiency. You are giving it, you are using up the iron, so there is less iron. You get to the iron-restricted erythropoiesis if you keep on giving ESA. And this is the inflammation-induced iron deficiency, where you have the storage iron but you are not able to release it. You’re not able to absorb it; so, ultimately, you’re left with less iron for actual erythropoiesis.
Slide 26
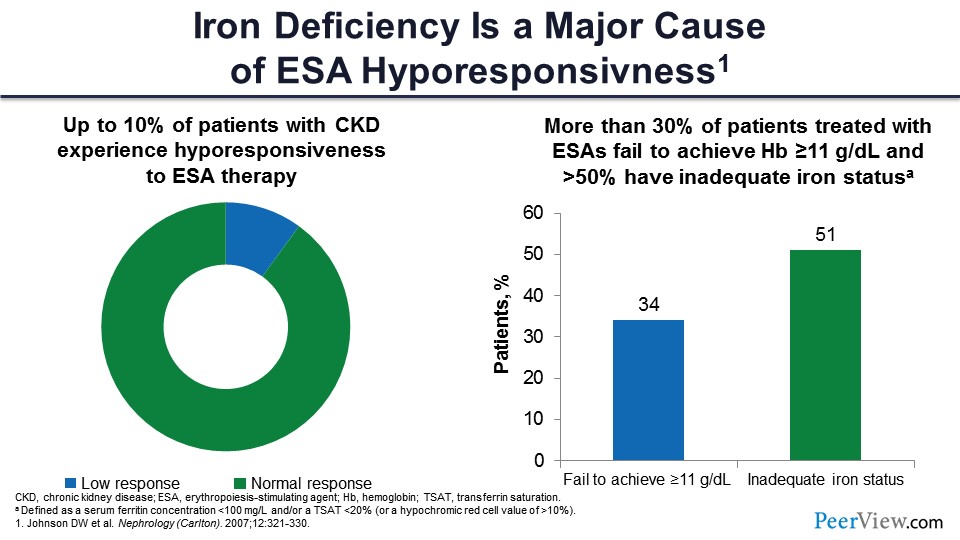
Let’s talk about ESA resistance. Say we are giving a lot of epoetin alfa, and it’s just not working. That’s a common condition; about 10% of patients in the United States have hyporesponsiveness to ESA, usually defined as more than 300 U/mL/week of epoetin alfa given subcutaneously. The hemoglobin is not able to go up over 11 g/dL despite that. That is called ESA hyporesponsive.
And about 30% of patients who are on good dose of epoetin alfa fail to achieve hemoglobin more than 11 g/dL, and 50% of those people are actually iron deficient. Iron is extremely important to the erythropoiesis process, and you need to ensure that there is not only storage iron, but that it is also available. But we haven’t had tools to release that iron until now.
Slide 27
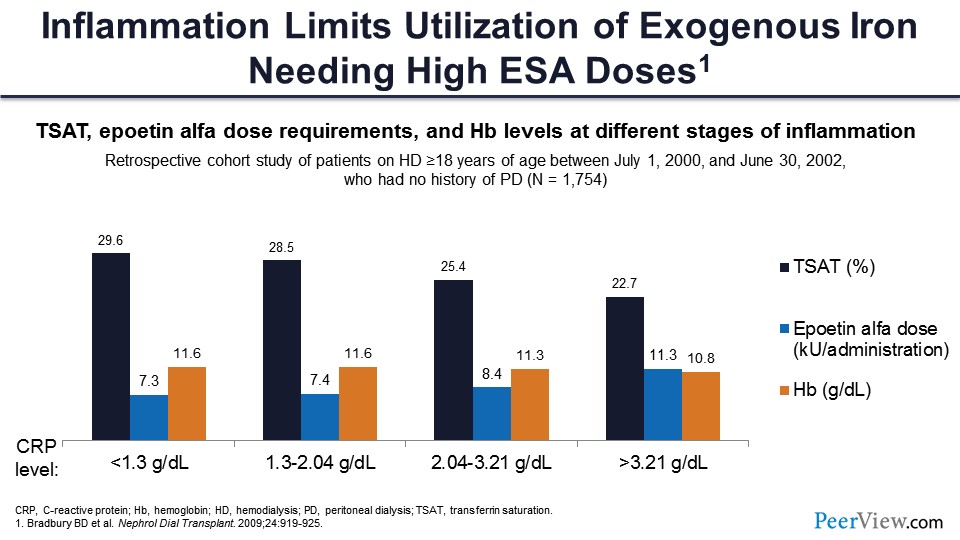
This is the scenario in inflammation. There are three things: We are checking TSAT in black, the ESA dosage in blue, and the hemoglobin in orange. As the CRP level goes up (an indicator of inflammation), the TSAT starts going down, the hemoglobin starts going down, and the ESA starts increasing. And that’s when you are trying to give a high dose of epoetin alfa to raise the hemoglobin to a high target. That’s where you get in trouble.
Slide 28

To summarize my part: CKD anemia is multifactorial, as we all know. It’s primarily because of erythropoietin insufficiency, but there is a component of iron deficiency, and there is a big component of inflammation that was not recognized until very recently. The current paradigm of just giving erythropoietin, just giving iron was okay, but it was not good, and there were consequences, dire consequences. There were major cardiovascular events associated with high dosages of erythropoietin and high doses of iron, trying to get hemoglobin to a certain level. And that was proved by RCTs. And that’s why we need new methods of treating anemia. And I will invite Dr. Wish, chair of this symposium, to tell you about those. Thank you very much.
MasterClass #2: Expert Insight on the Clinical Potential of HIF-PH Inhibitors in CKD-Associated Anemia
Slide 29
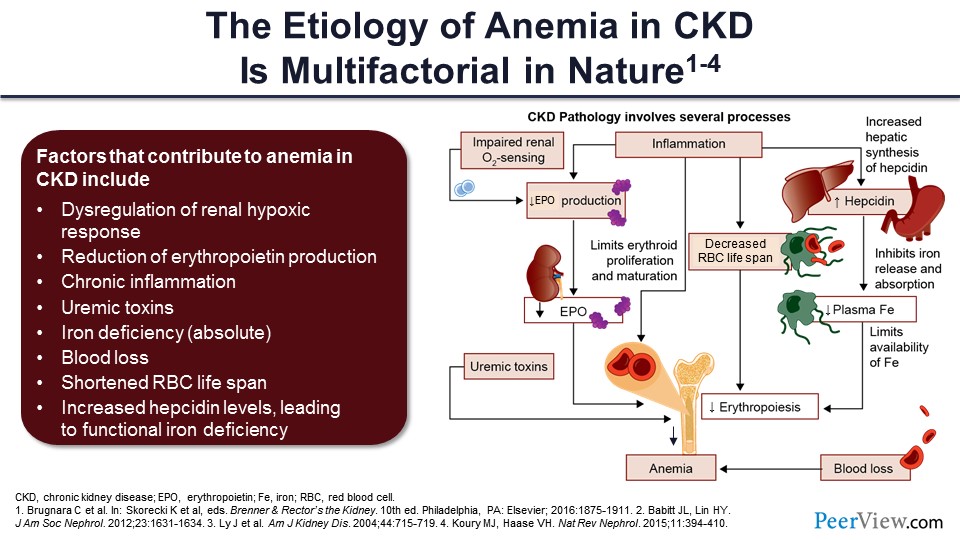
Dr. Wish: Thank you, Anil, for an outstanding presentation and for laying the groundwork for what I’ll be covering in the next 25 or so minutes: the new approaches to anemia that address the issues of inflammation and functional iron deficiency. A lot of what I’ll be presenting to you in terms of oxygen sensing and the anemia of CKD was the basis for awarding the Nobel Prize in Medicine and Physiology this year. For those of you who keep track of Nobel Prizes, three individuals, one of whom was a nephrologist, won the Nobel Prize for this very work that I will be presenting during the next 15 or so minutes.
As Anil pointed out, CKD anemia is multifactorial. We talked about the reduction in erythropoietin production and the chronic inflammation. There is also the role of uremic toxins that downregulate erythropoiesis and also decrease red cell lifespan. Some patients have absolute iron deficiency, but the majority has the functional iron deficiency that Dr. Agarwal explained. There is blood loss because of decreased platelet function, etc. We mentioned the shortened red cell lifespan, and Dr. Agarwal elegantly presented the issue with the increase in hepcidin levels, which lead to functional iron deficiency. So, I’ll be adding to this. The first bullet point, which is the dysregulation of the renal hypoxic response, and basically what you see on the right-hand side of the slide in the diagram, is all of these kind of put in order in terms of how they ultimately result in anemia.
Slide 30
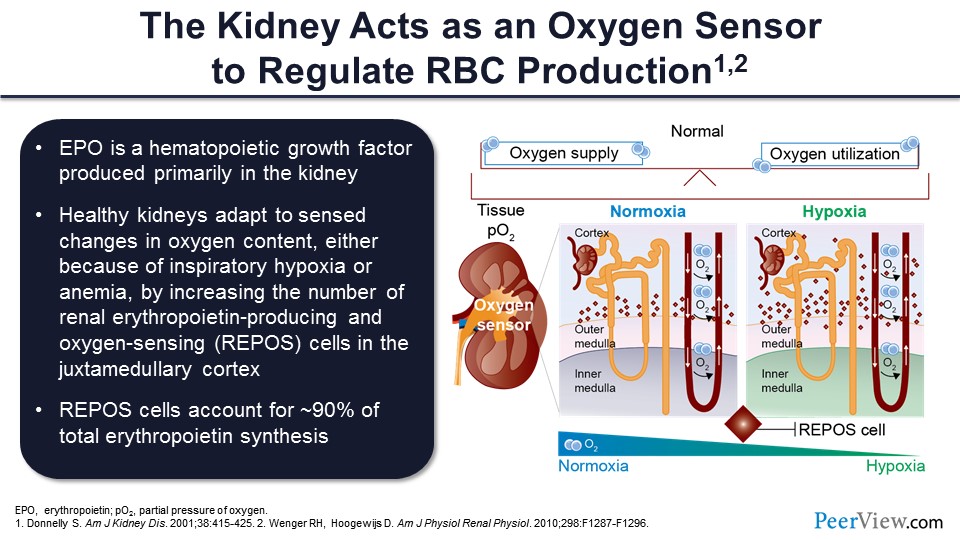
As it turns out, the kidney is the major oxygen sensor that the body uses to regulate red blood cell production. You would think: Why the kidney? And the reason is because the kidney is an extremely oxygen-consuming organ. When you think about it, a normal kidney or normal pair of kidneys—and hopefully your kidney function is close to normal—filters about 180 L a day of plasma, and yet you only make 2 L a day of urine by and large.
So, the kidney is reabsorbing 99% of that filtrate. And to reabsorb 99% of almost 200 L of filtrate is an extremely energy-dependent process that requires the delivery of lots of oxygen. So that is why nature chose the kidney as the oxygen sensor because any disruption in the oxygen supply and demand balance can have adverse consequences. The body tries to overcome that imbalance by making more red cells, so that more oxygen can be delivered to the kidney and also, of course, to other organs like the brain and the heart.
Healthy kidneys will adapt to sensed changes in oxygen content, either because of inspiratory hypoxia, if you live at a high altitude or you have COPD, by increasing the number of these renal erythropoietin-producing, oxygen-sensing cells, which are REPOS, in the area of the kidney that’s called the juxtamedullary cortex. This is the cortex of the kidney that is very, very close to the medulla—kind of the interface between those two layers of the kidneys.
And as it turns out, REPOS cells account for about 90% of total erythropoietin synthesis, and the rest of the 10% that is not in that 90% actually comes from the liver. And this explains why, as you will see, even patients who are anephric, who have no kidney function at all, if you give them drugs that simulate a hypoxic situation, which is what the class of drugs that we’ll be talking about does, you can actually get adequate erythropoietin production outside the kidney from the liver.
On the right-hand side of the slide, those little diamonds in the juxtamedullary cortex represent these REPOS cells. In the setting of hypoxia, the number of these cells increases so they can produce more erythropoietin, and that increased amount of erythropoietin could stimulate the bone marrow to make more red blood cells and deliver more oxygen to the kidney, so the kidney is no longer bathed in a hypoxic environment, if you will.
Slide 31
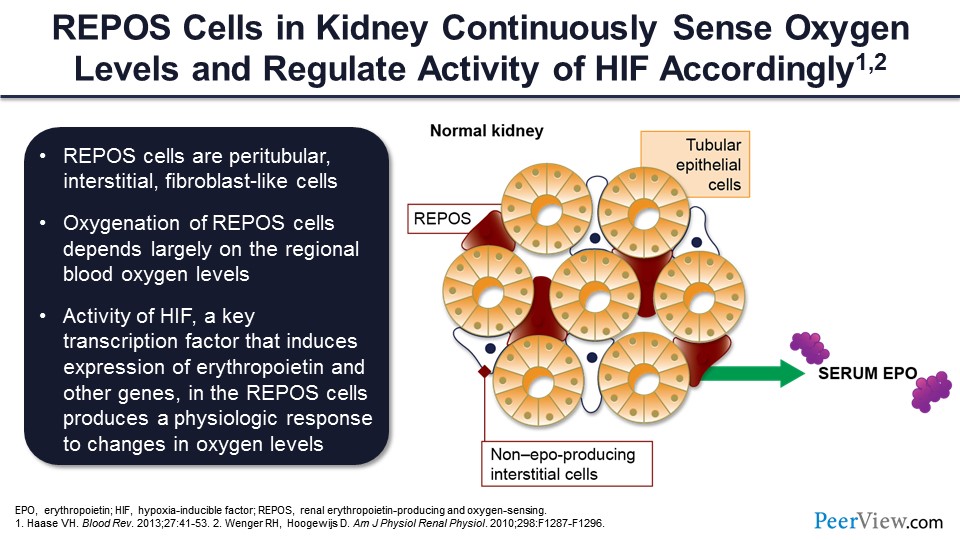
So, you have oxygen sensing, and then the intermediate step is the production of this protein called hypoxia-inducible factor, one of the effects of which is to stimulate the genes that produce erythropoietin, which allows for erythropoietin to increase bone marrow red cell production, which then again feeds back to these hypoxia-responsive cells, more oxygen delivery, then a downregulation, and the achievement of a new steady state.
Slide 32
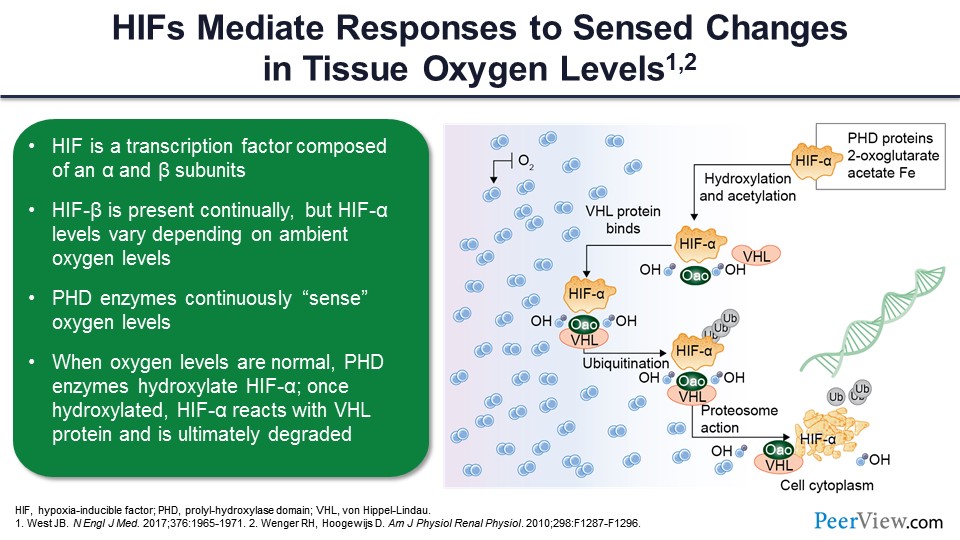
So HIFs ultimately mediated the responses to these sense levels or sense changes in tissue oxygen level. Now, as it turns out, HIF contains two subunits: HIF-α and HIF-β. HIF-β is produced continuously. There’s always HIF-β around, but HIF-α varies in its activity based on the presence of oxygen. If oxygen is present, then there is a group of enzymes, called prolyl-hydroxylase domain enzymes, which continuously sense the oxygen and use the oxygen as a cofactor for their activity. And if oxygen is present, these enzymes become active and degrade HIF-α so that you basically have no HIF dimerization and no stimulation of erythropoietin, and ultimately, this can cause anemia. So these HIFs coordinate a series of responses to hypoxia that regulate erythropoiesis, as we will see, not only through their ability to stimulate erythropoietin but also their ability to change iron metabolism. In hypoxic conditions, the prolyl-hydroxylase domain enzymes are not activated. This prevents the HIF-α from being degraded.
If oxygen is present, then as you can see on the right-hand side of the slide, the prolyl-hydroxylase enzyme—basically hydroxylase—is the proline residues within the HIF-α, and this changes the configuration of the HIF-α so it is sensed by the enzymes, which ultimately degraded, one of which is von Hippel-Lindau factor. This leads to the degradation of alpha, and no production of erythropoietin. In the absence of oxygen and the inactivity of the prolyl-hydroxylase enzymes, the HIF-α can dimerize with HIF-β, go into the nucleus, and ultimately, it is going to exert its effects to stimulate a variety of genes that code for a number of proteins not only involved in red cell production but also involved in many other ways that the organism uses to adapt to a hypoxic environment.
Slide 33
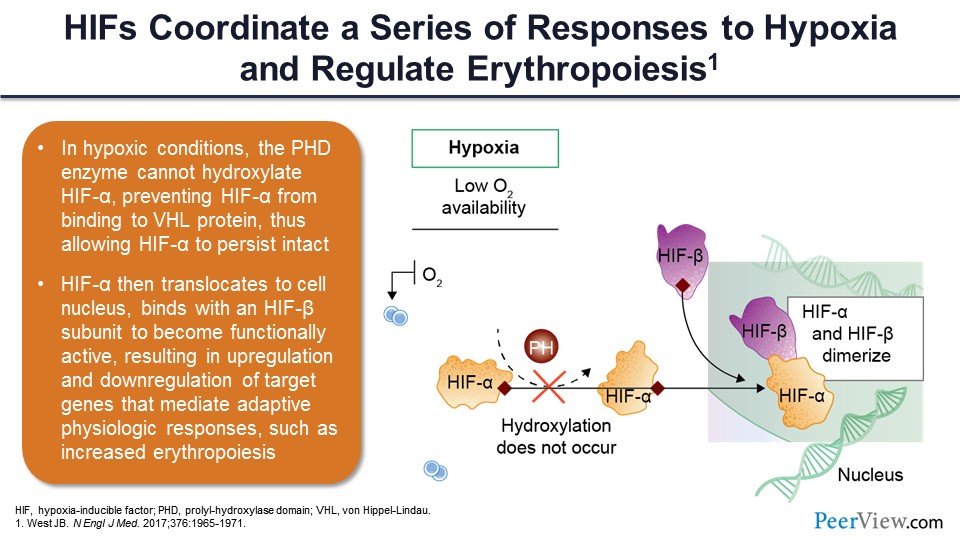
You can see here in a kind a schematic way that if you have hypoxia moving from left to right, low oxygen availability, and also normal amounts of oxygen consumption, this causes increased iron mobilization and absorption, increased iron transport to bone marrow, increased erythropoiesis, and then ultimately the target genes are affected. The increased iron absorption and mobilization through the duodenum is mediated by two factors: DMT1, which is divalent metal transporter-1, and duodenal cytochrome B. This allows the iron to traverse from the GI lumen through the duodenal enterocytes and into the plasma, where it combines with transferrin.
Slide 34
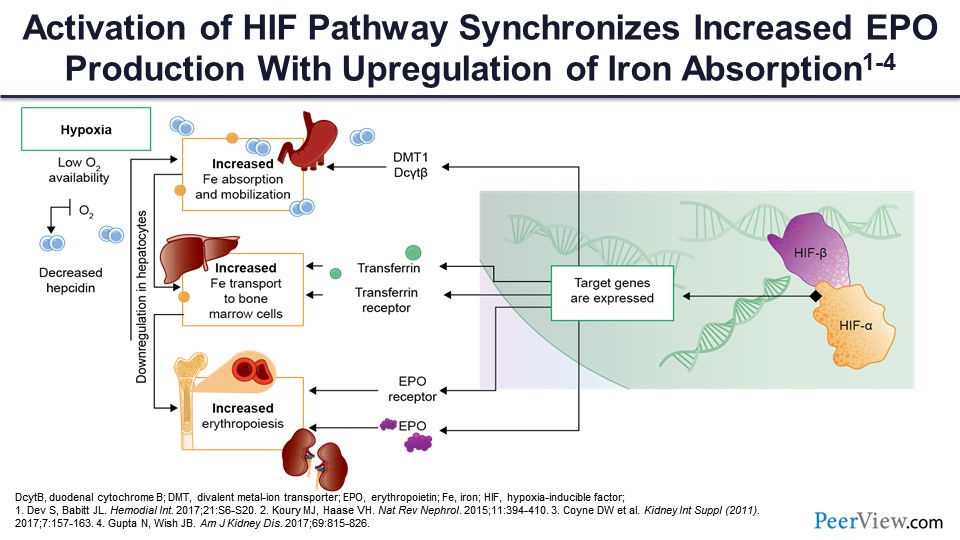
Increased iron transport to the bone marrow cells is mediated through increased production of the transferrin carrier protein, as well as increased expression of the transferrin receptor on the erythropoietic precursor cells. So basically, what you have to do is not only pitch the iron from the transferrin but develop a catcher’s mitt so that that iron can be captured by the erythroid precursors incorporated to the cell to support hemoglobin synthesis.
And finally, as we mentioned, one of the important effects here is not only the increased production of erythropoietin but also the increased expression of the erythropoietin receptor on the stem cells and on the early erythroid precursor cells—the burst-forming units and the colony-forming units that you saw on the previous slide—so that increased levels of erythropoietin can be sensed by these cells and ultimately exert its erythropoietic effect.
Slide 35
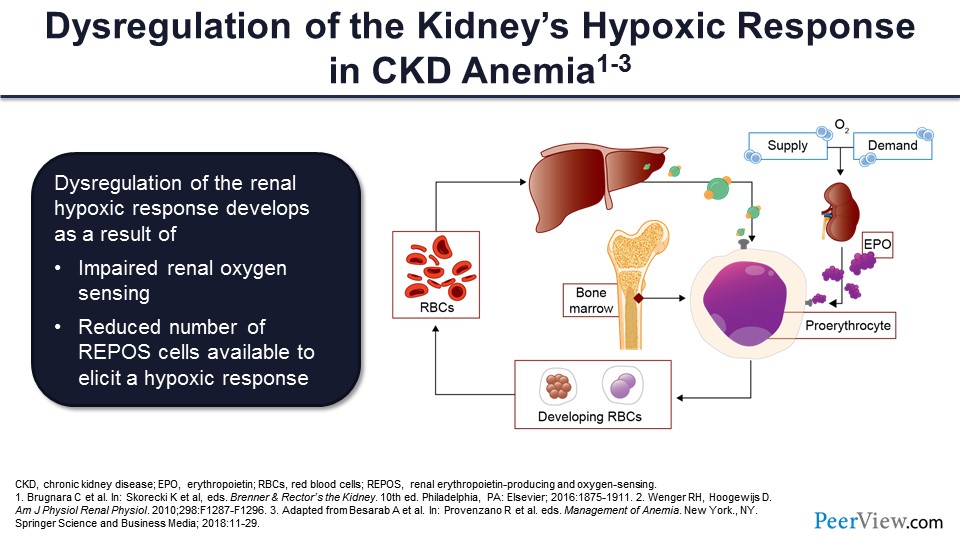
In CKD, you have this dysregulation of the kidneys’ hypoxic response to anemia. There’s impaired oxygen renal sensing, as well as a reduced number of these renal erythropoietic-producing cells, and this combines to decrease erythropoietin production and red cell supply.
Slide 36
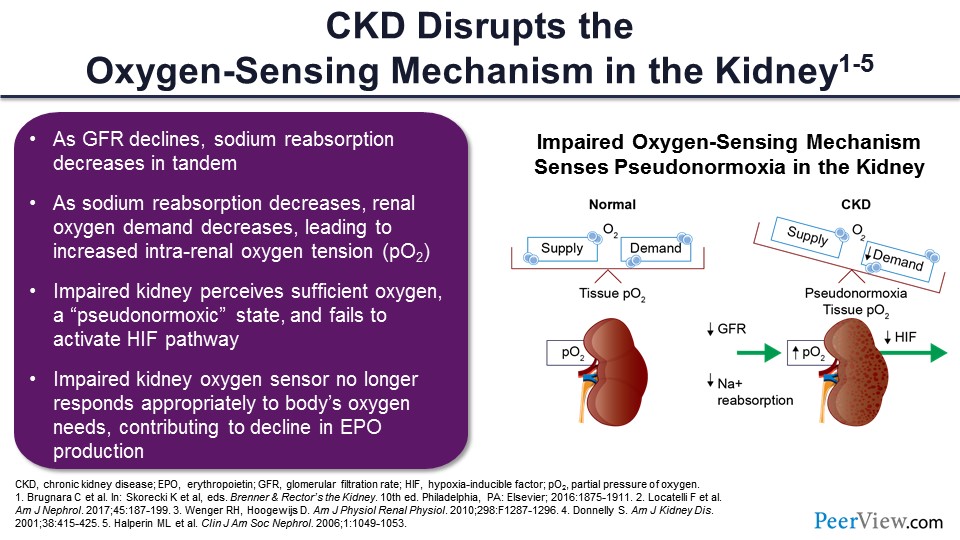
If you move to the extreme right-hand side of this slide, it’s ultimately a function of oxygen supply and demand, and we’ll show what’s going on here. In the setting of CKD, the tubules are sick. The tubules are not reabsorbing filtrate for the same degree as in normal individuals. You have fewer tubules functioning, and as a result, you have altered supply and demand.
Even in the presence of normal supply, since demand goes down even as hemoglobin goes down, the kidney doesn’t get it. If demand is decreased and supply is increased in the setting of the anemia, the kidney, the REPOS cells think that everything is fine, and they don’t secrete erythropoietin and otherwise stimulate red cell production. So again, the sick kidney is fooled into thinking this supply–demand balance is okay because the demand goes down to the same extent as supply goes down.
Slide 37
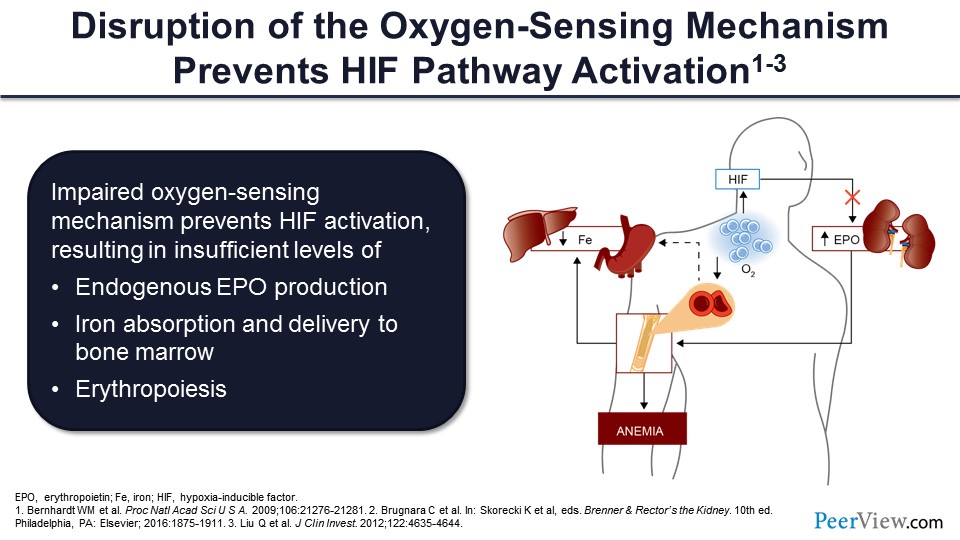
This disruption in oxygen-sensing mechanism prevents HIF pathway activation, decreasing endogenous erythropoietin production, and ultimately decreasing erythropoiesis.
Slide 38
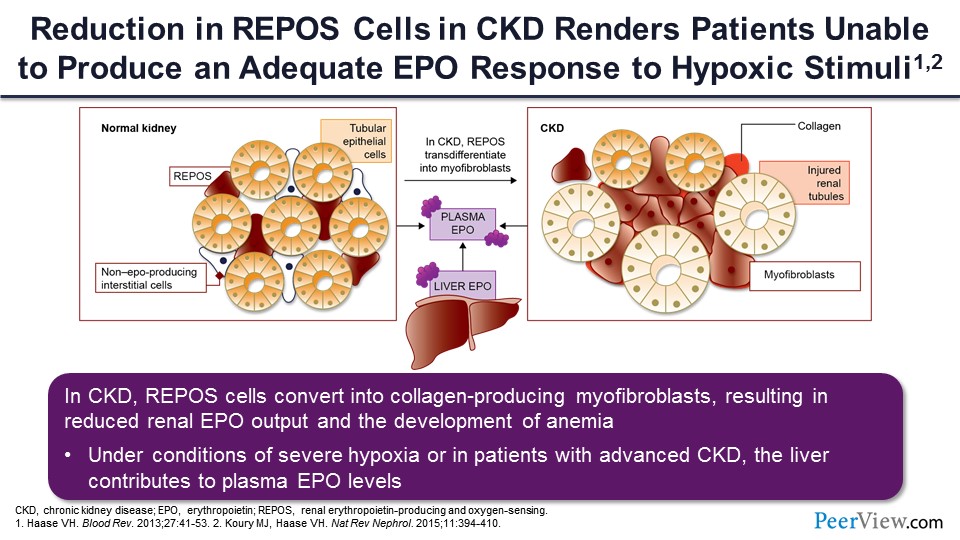
What you see here is kind of a histologic representation of what goes on. In the left-hand side is a normal kidney where those little kind of triangular cells in between the tubules represent the REPOS cells. As the kidney gets sicker and the patient gets more in the way of tubular atrophy and interstitial fibrosis, which is a hallmark pathologically of more advanced CKD, you can see there’s a lot of transformation of these REPOS cells into non–REPOS-producing myofibroblasts, and this shifts the burden of erythropoietin production from the kidney to the liver. Okay, so the liver takes on more of a role; but remember, under normal circumstances, the liver is only producing 10% of erythropoietin. So even if the liver were to step up and produce 20%, the reduction in kidney production of erythropoietin is so profound that the liver cannot possibly fill that void.
Slide 39
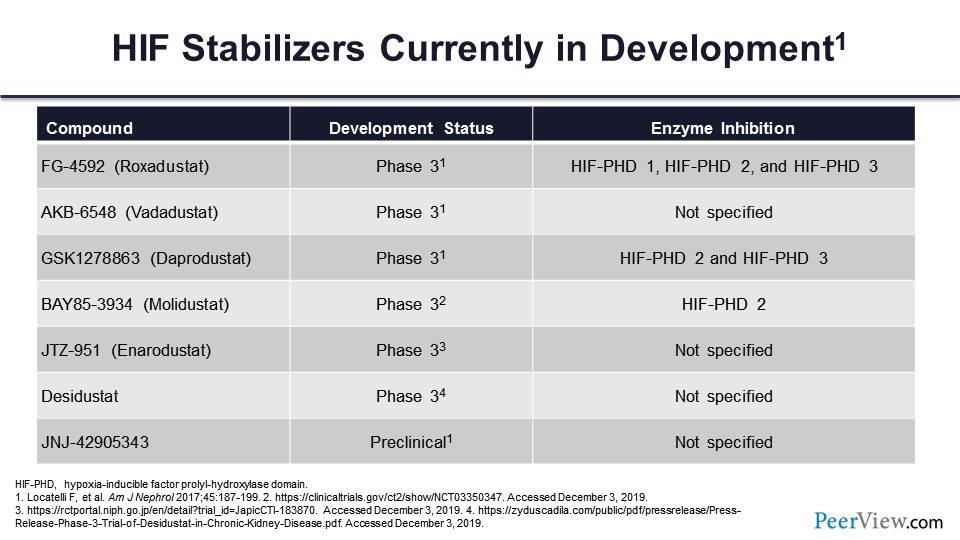
We have a new class of drugs called HIF stabilizers or HIF prolyl-hydroxylase inhibitors, which basically inhibit that enzyme, prolyl-hydroxylase, just as if oxygen were absent. And this allows for the survival of the HIF-α—prevents its degradation—so it can combine with HIF-β and ultimately stimulate a variety of genes that are related to the hypoxic response, including but not limited to the changes in erythropoietic hormone and iron metabolism that we’ve already mentioned.
The first three are the ones that are currently under development in the United States: roxadustat, vadadustat, and daprodustat. They’re all in or have completed, in the case of roxadustat, phase 3 trials. Going from molidustat on down are four agents that are being developed in other parts of the world; again, many of these are also in phase 3 development.
Slide 40
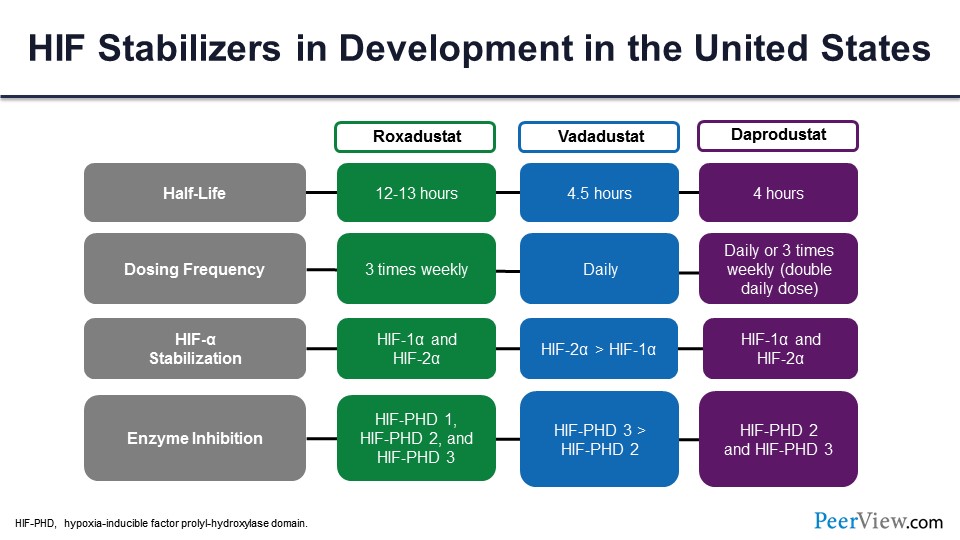
So let’s concentrate on the three that are in development in the United States. You can see that there are some subtle differences in terms of their pharmacokinetics. Their mechanism of action (MOA) is identical; they all basically inhibit those prolyl-hydroxylase domain enzymes, allowing for more HIF-α and ultimately more effect of this hypoxic stimulus.
Roxadustat has the longest half-life: 12 to 13 hours. As a result, it can be administered three times weekly. It seems to have more of a HIF-1-α and HIF-2-α effect. And as you can see, the enzyme inhibition for prolyl-hydroxylase domain (PHD) is 1, 2, and 3.
Vadadustat has a shorter half-life of 4.5 hours and usually is given daily, at least in the phase 3 clinical trials. All of them use it daily. It seems to have a greater effect on HIF-2-α than HIF-1-α, and the prolyl-hydroxylase seems to be more PHD 3 versus PHD 2.
Daprodustat has a similar half-life as vadadustat: 4 or so hours. It can be given daily, but there’s been a phase 2 study that shows that it’s effective when given three times weekly at double the daily dose. It affects HIF-1-α and HIF-2-α, and it affects prolyl-hydroxylase (PHD) 2 and 3.
Slide 41

So, what do we have in terms of completed phase 3 studies? And the answer is not a lot. The one that has been published in a peer-reviewed journal—actually two companion pieces—are roxadustat for anemia in patients undergoing dialysis and roxadustat for patients not undergoing dialysis—two separate articles that appeared in the New England Journal of Medicine in July of 2019.
Slide 42

Basically, what they found in these studies of 6 months or less—they were not 3-year studies, and they were not adequately powered for long-term cardiovascular events—is that in nondialysis-dependent patients, it was superior to placebo. In dialysis patients, it was noninferior to ESAs, it was not adequately powered for MACE outcomes, and there were MACE. There was increased hyperkalemia in both nondialysis-dependent and dialysis patients on the active drug. There were no other safety signals.
Slide 43
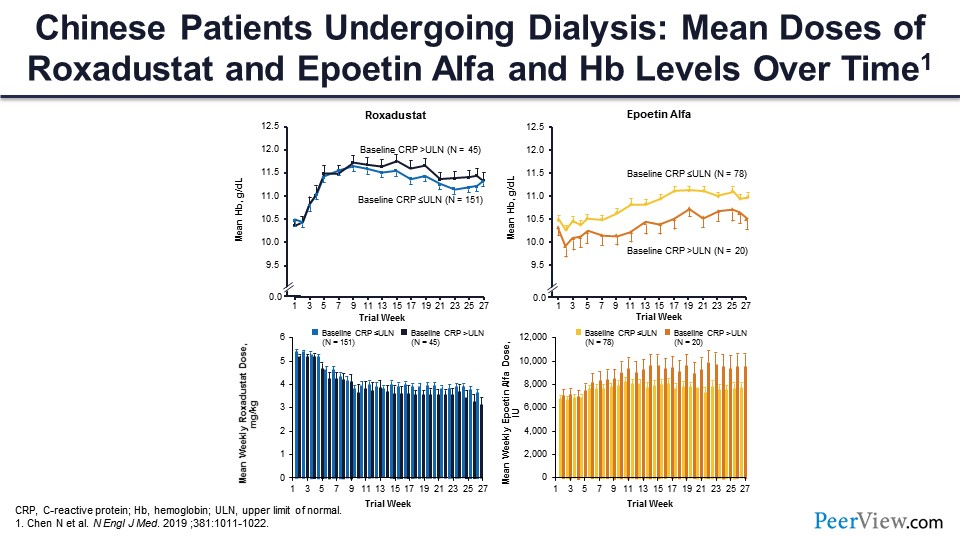
And what I found to be most interesting is that it was equally effective in patients with and without inflammation based on their CRP levels in the hemodialysis study. So this is the figure that appeared in the article. The patients treated with roxadustat are on the left-hand side of the slide, and you can see the patients who had a high CRP—upper limit of normal, greater than upper limit of normal—and patients who had a normal CRP, and the hemoglobin response was identical in the two groups. The roxadustat dose that was required to achieve that hemoglobin response was identical in the two groups. So, roxadustat seems to be inflammation blind in terms of its effectiveness. On the other hand, with ESA, which was in the control group in the dialysis population, you can see there was a significantly lower hemoglobin response among those patients with high CRP, and a higher ESA dose was required among the patients with the higher CRP, much as we’re used to in clinical practice now.
Slide 44
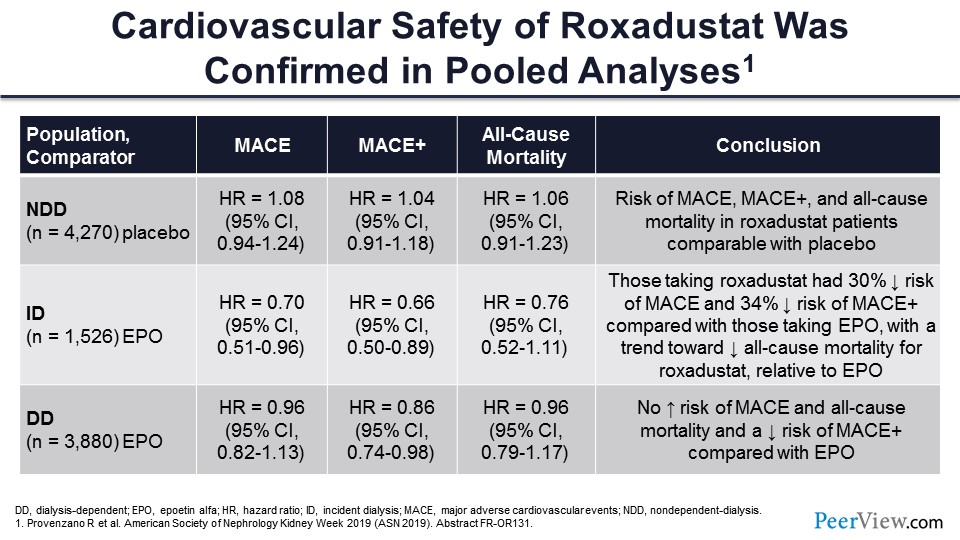
So the cardiovascular safety has been confirmed in the pooled analysis of the phase 3 studies that have not yet been subject to peer review. In nondialysis-dependent patients, MACE and MACE+ were comparable with placebo, so that’s very, very important. These drugs in the nondialysis-dependent patients have no worse effects in terms of cardiovascular endpoints than placebo.
In incident dialysis patients, those taking roxadustat actually had a decreased risk of MACE and MACE+ and a trend toward decreased all-cause mortality compared with erythropoietin. And in the dialysis-dependent patients—so this is the pooled incident and prevalent dialysis population—there was no increased risk of MACE or of all-cause mortality and actually a decreased risk of MACE+.
So if you’re looking at efficacy, it’s very, very clear that these drugs are better than placebo, comparable with ESAs in patients without inflammation, and probably superior to ESAs in patients with inflammation. But the “but” is: What about safety? And there you have very, very attractive results, as well. No worse safety than placebo in nondialysis-dependent patients, better safety in incident dialysis-dependent patients, and comparable safety with ESAs in overall dialysis patients.
Slide 45

There have been a number of other unexpected effects of roxadustat in these pooled phase 3 analyses. There’s a slower rate of EGFR decline in nondialysis-dependent patients, the mechanism of which is not well understood. These are topline results. Improvement in quality of life measures in nondialysis-dependent patients as compared with placebo. And as I showed you, the efficacy irrespective of the inflammation status.
Slide 46
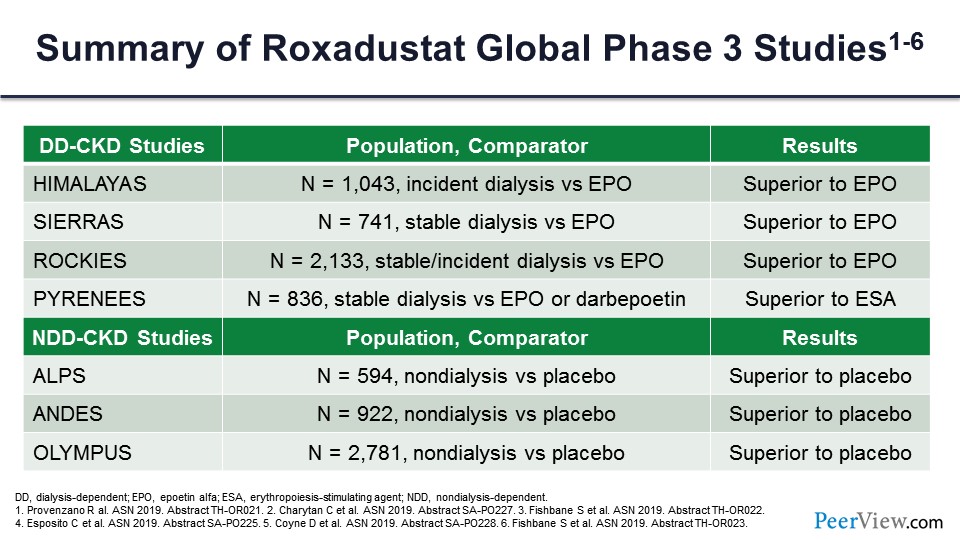
This is a summary of the roxadustat global phase 3 studies. You can see again: consistently superior to erythropoietin in the dialysis-dependent studies in terms of efficacy; and in the nondialysis-dependent studies, consistently superior to placebo in terms of efficacy.
Slide 47

Vadadustat is kind of the second in line in terms of the timing of its phase 3 clinical trials and ultimate approval by the FDA. It’s estimated that roxadustat—if everything goes well—will probably be approved by the FDA maybe a year from now—fourth quarter 2020—and vadadustat may be a year behind. What we have is much smaller results of phase 2 studies. As you can see, not surprisingly, there’s comparable efficacy with ESAs. They did not do placebo-controlled trials in their nondialysis-dependent patients; they did ESA controlled, and efficacy and safety, so far, seem to be comparable with ESA.
Slide 48

These are the ongoing phase 3 trials of vadadustat. As you can see, they all have O2s in them to remind you that these have to do with oxygen sensing and basically inhibiting what would otherwise be the oxygen effect of prolyl-hydroxylase. And again, the comparators are primarily ESAs in all the groups.
Slide 49

Daprodustat, one more year behind, probably won’t be approved, again if all goes well, until 2022. Again, these are much smaller, phase 2 studies.
Slide 50

But they also had a very large phase 3 program, which are all variations of this ASCEND model. And again, as you can see, these are all ESA control studies except for the ASCEND-NHQ study, which is versus placebo in nondialysis-dependent patients.
Slide 51

Other potential beneficial effects include reduction in cholesterol; reduction in hepcidin, which we already talked about; and even reduction in blood pressure.
Slide 52
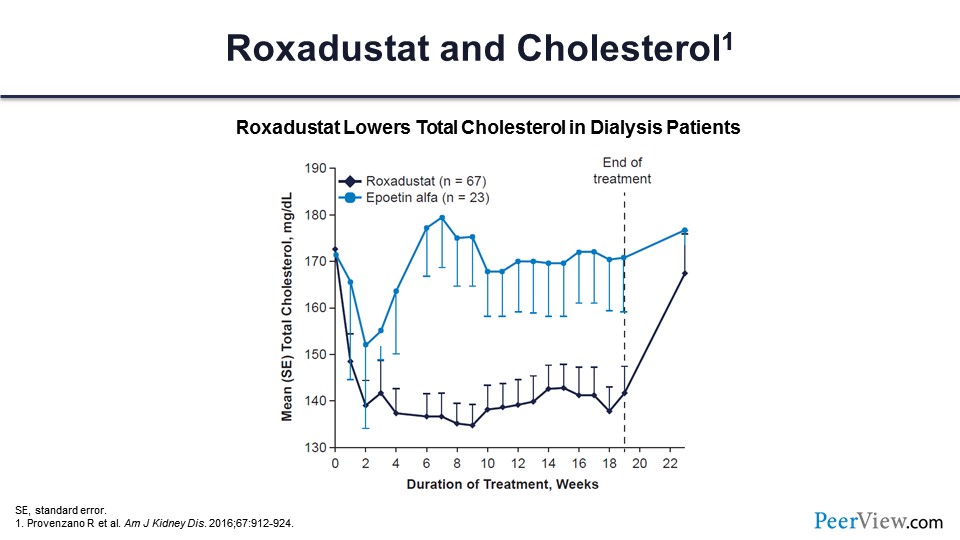
This is roxadustat and cholesterol. You can see an average reduction in cholesterol levels of about 30 mg/dL compared with placebo.
Slide 53
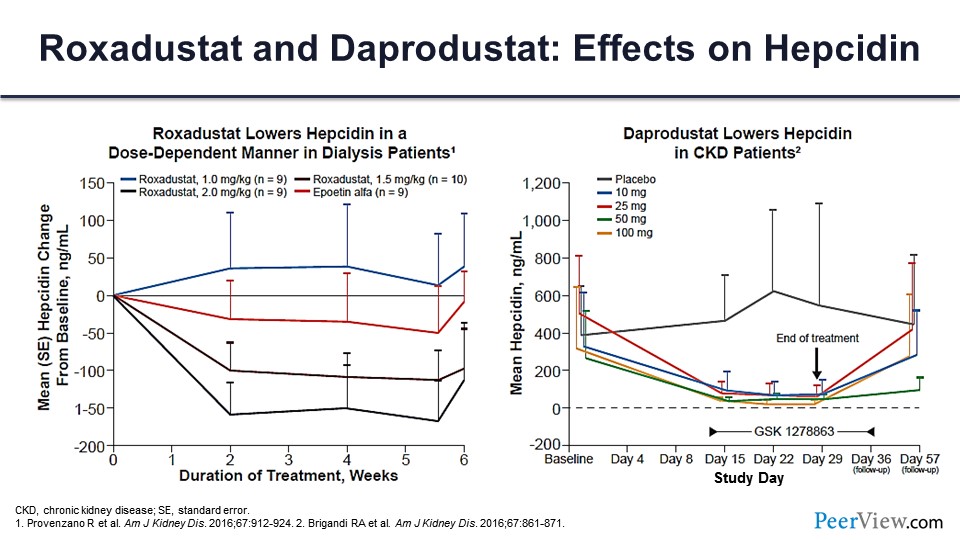
This is the effects of daprodustat on hepcidin, which you can see. With roxadustat, what you can see is more or less dose related.
Slide 54
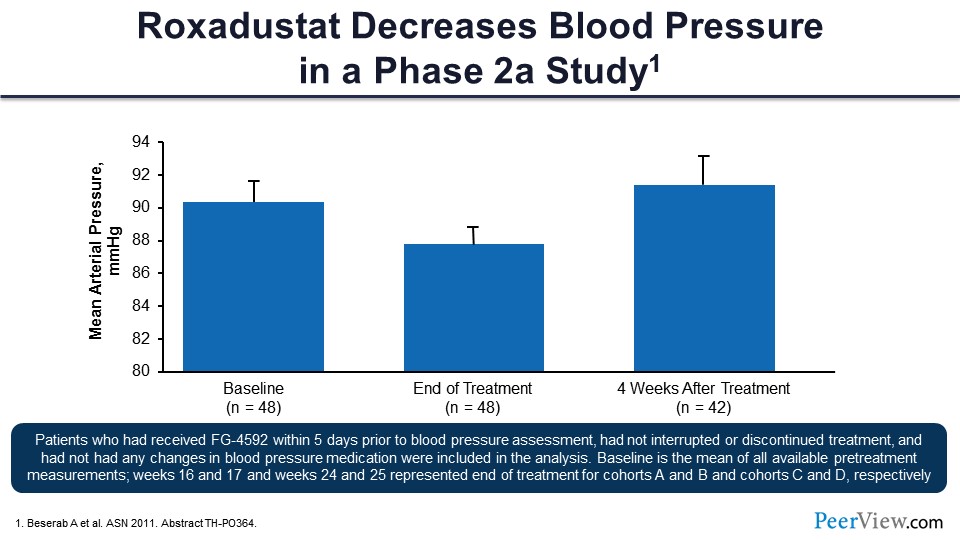
And this was one very, very small phase 2 study of roxadustat on blood pressure that showed about a 5-mmHg decrease in mean arterial pressure compared with baseline.
Slide 55

The safety concerns have to do with the fact that the HIF stabilizers not only stimulate the genes related to erythropoietin and iron mobilization that we talked about, but they stimulate hundreds, if not thousands, of other genes that are related to the systemic response to hypoxia, including glucoregulation, angiogenesis, extracellular matrix, cell proliferation, and survival.
Slide 56

So far, we have not seen any of these safety concerns play out in the larger phase 3 trials. As we said, no increase in blood pressure, maybe even a decrease. Possible association with pulmonary hypertension, but again, this is more theoretical than evidence based. No increase in VEGF levels—which is the angiogenic factor, that vascular endothelial growth factor—so, that somewhat allays the concerns, at least in the short term, that you’re going to see more angiogenesis that may lead to tumor growth or diabetic retinopathy, for example. And again, unfortunately, the degree of statistical power that we have so far in terms of the phase 2 and phase studies limits our ability to assess these.
Slide 57

You ultimately have to consider the value proposition of these drugs, and I think the value proposition is primarily going to be in patients who are ESA failures. You need a better mousetrap, and now we have a class of drugs that does seem to be effective in patients who require higher ESA doses or don’t achieve ESA targets.
Slide 58

And I think in the nondialysis-dependent population, these are orally administered drugs. So from a patient perspective, it’s going to be a lot more attractive for them to take an oral drug daily or three times a week than to have to self-inject an ESA, a darbepoetin, an erythropoietin every week or every month, or have to go to a clinic to get that injection. And again, I think from a patient standpoint, much more user-friendly.
Slide 59

And just to summarize, HIF stabilizers are, in fact, the first revolutionary treatment of CKD since ESAs were introduced 30 years ago. They may be more attractive and even replace ESAs in nondialysis-dependent patients because they’re oral, and they’re no less safe than placebo. And they may actually be ordered by non-nephrologists who are somewhat, I guess, scared of the black box warning that you have with the ESAs. They are effective in patients who are ESA-resistant with inflammation. The value proposition remains to be seen. So that concludes my comments.
Practicum: Defining the Role of Pharmacists in the Evolving Treatment Landscape for Anemia in CKD
Slide 60
Dr. Dowling: Thanks, Dr. Wish and Dr. Agarwal for reviewing the pharmacology, MOA, and the various issues around this brand-new class of drugs. The question now becomes what is the role of the pharmacist going forward in this kind of evolving picture of anemia treatment and anemia management, specifically in the CKD population.
I’m going to review some of that in terms of pharmacy. What is our role going forward in anemia management? There’s been a lot done within the nephrology pharmacy specialty, specifically within the American College of Clinical Pharmacy (ACCP) Nephrology Practice and Research Network (PRN). A number of individuals have been really forging the frontier in terms of anemia management. I’m going to highlight some of the activity that’s been done by pharmacists already in this area, but there’s just really a growing role for pharmacists in this area, as these new anemia treatments are emerging.
So I’m going to review some of the basic background in terms of the pharmacist’s role in CKD management. Again, the Nephrology PRN has been very active in that. I had the great opportunity to serve as chair of the Pharmacist Nephrology Group a number of years back, and this was really a focus of that group.
But I’ll just review some of the activity of pharmacists in the CKD space right now and some of the components going forward related to collaborative practice agreements, where really a lot of this, the decision algorithms and things, will come into those collaborative practice agreements (CPAs) going forward, and pharmacists will have a role in helping to maximize the care of those patients. And lastly, we have a couple of cases that really kind of wrap up some of the concepts, as well, that we’ve been talking through.
Slide 61

There have been a number of papers published in the peer-reviewed literature describing the role of the pharmacist in multidisciplinary CKD clinics, some of them involving anemia management specifically, but in some cases really more general practice of pharmacy. And this slide shows data from a paper published in Nephrology Dialysis Transplantation that was a review of pharmacist activity, demonstrating a number of different models that were being described in the literature.
The paper was a systematic review, really kind of an international group that looked at this—37 different studies in more than 4,700 patients, where pharmacists were involved in the care of patients with CKD. Of these 37 studies, eight of those were controlled studies, where there was a pharmacist group and then some other usual care group—pharmacy physician extenders and things like that.
But eight of those studies were controlled. In just over 700 patients, they found some trends in significant benefits to the patients in terms of quality of care, some reduced composite endpoints in end-stage renal disease (ESRD) and mortality in some subpopulations like those with diabetes, and reduced all-cause hospitalizations, as well as improved anemia management in the pharmacist-managed CKD clinics. Primarily, those patients received or achieved the target hemoglobin, which was a range of 10 to 12 g/dL in a higher upper proportion. Four of the studies in this systematic review had evaluated health outcomes, health QOL, and did have some improvements, as well, in the pharmacist groups that had measured those QOL indices.
So these are data to suggest the most comprehensive review of pharmacist activities in CKD, particularly anemia management, as well, that showed a significant impact of pharmacists in the care of these patients and particularly with collaborating with our physicians in terms of care.
Slide 62
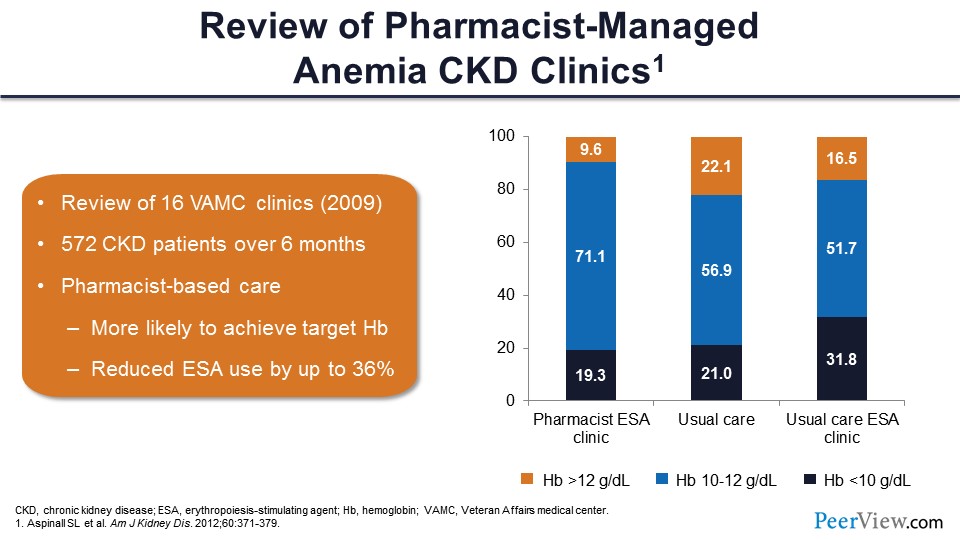
This slide shows a couple of studies that looked at anemia management specifically in CKD. This is a study that outlines some of the benefits of having a pharmacist in an anemia management clinic, specifically. This was Sherri Aspinall’s group at Pittsburgh, and they looked at a number of Veterans Affairs medical center clinics across the United States. They had 16 centers that had pharmacists involved in CKD clinics that had anemia management, and they compared the groups where pharmacists were involved with other usual care groups. A few of the centers had usual care in an ESA clinic, where a PharmD was not present.
They found really good outcomes with the pharmacist-based care. They found that those patients were more likely to achieve a target hemoglobin; again, that was in the 10 to 12 g/dL range, about 72% within that range. Fewer patients were above the 12 g/dL cutoff, and fewer were below the 10 g/dL as well. They found that pharmacist-based clinics were associated with a better likelihood to achieve a target hemoglobin, and there was reduced ESA use within those groups by almost 40% or so. So, there were really good outcomes in the pharmacist care clinics with anemia management.
Slide 63
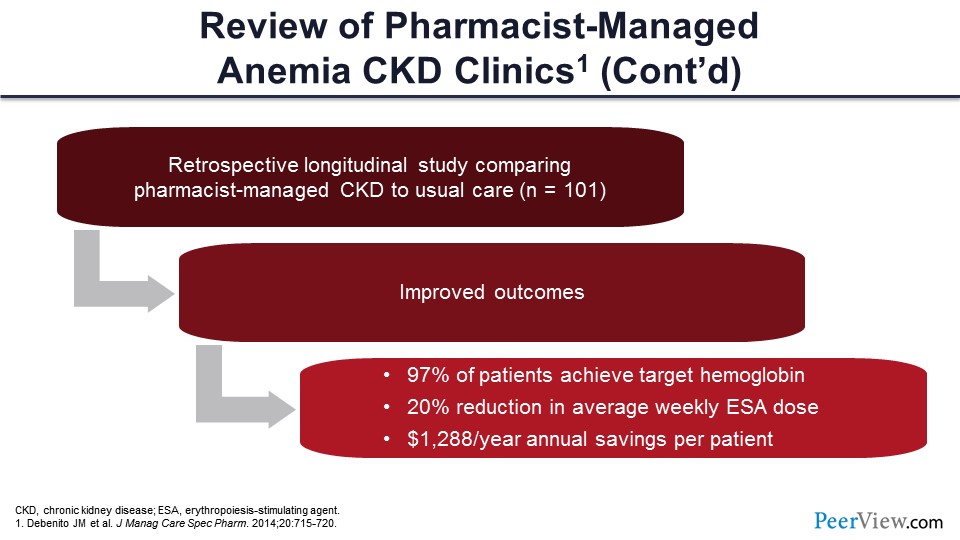
This slide shows another paper published on general managed care specialty pharmacy. This paper looked retrospectively at a group of patients with CKD managed by pharmacists compared with usual care—just about 100 patients here. Again, they found improved outcomes in those patients who had a pharmacist involved in their care: 97% of patients achieved a target hemoglobin, there was a 20% reduction in average weekly ESA dose, and a cost savings in terms of the pharmacoeconomics of approximately $1,300 per year in annual savings based on ESA use.
Slide 64

And the last example: This slide shows a case management series that was published by Hugh Easley’s group in Montana that looked at a significant need to incorporate a pharmacist into the ESA and anemia management clinics within their health system. There was a need from the physicians and nephrologists to help with the care of those patients, and so they started a clinic de novo and described that as part of an American Society of Health-System Pharmacists’s Practice Advancement Initiative (ASHP PAI) back in 2017.
And so, they nicely laid out the development of this type of a clinic. The title of that was “Pharmacist-Managed Anemia Clinic Improves Guideline Adherence for Darbepoetin”; but in their analysis, they did a 12-month pre-evaluation and 12-month post-evaluation of their pharmacist-developed clinic. In their clinic, they had two ambulatory care pharmacists. We’ll see this as a common model—a sharing model between an anticoag pharmacist who comes across to help with nephrology in the ESA clinics as well. So, that’s kind of a common model that you see, a sharing of ambulatory care pharmacists there.
In their outcomes, they saw some pretty dramatic shifts in terms of doses of ESAs being compliant in terms of guidelines. They saw a significant improvement in that activity. They also asked their physicians while they were developing that clinic how they were really satisfied with the activity, and they showed improved physician satisfaction with that particular clinic and their activities. They were very satisfied with the activity; and it also helped, in their view, to reduce the workload on the physician side in terms of being able to refer patients to their clinics and really have that time between a physician and the patients or the pharmacist and the patients in that setting.
They also reported improved Medicare reimbursement. In these cases, hemoglobin is measured within 7 days. There was improvement there, as well as with iron repletion prior to ESA, which is in the guidelines. There can be improvement there when pharmacists are involved in that type of care.
Slide 65

We know that there are a lot of data out there in the peer-reviewed literature on the role of pharmacists and how they’re really helping the care of the patients with CKD and specifically anemia. Now just to kind of operationalize that, I’m going to walk through a couple of examples of CPAs that you might consider in your practice settings.
CPAs can be general in terms of CKD management: Some manage multiple disease states but CKD is the qualifier for patients in that clinic. There are also CPAs that are specific to anemia management, so cases where the practice or the scope of practice is limited to anemia, as well as iron management.
Slide 66

I’ll walk through a couple of examples of these. One of the key components of the CPAs, in terms of anemia management in CKD, is certainly the state laws that define scope of practice within your state. I think the last time I looked, 48 of the 50 states allowed CPAs between a prescriber and providers that are designated by that prescriber. And so, it’s really a great opportunity for pharmacists to be involved in this multidisciplinary care.
Some of the CPAs actually go ahead and describe what a CPA is. It’s defined as a practice in which that prescribing practitioner makes a diagnosis, maintains ongoing supervision of the patient care, but also, at the same time, refers that patient to a pharmacist who then can either initiate modified drug therapies, as well as perform overall care of the patient under established protocols and often clinical practice guidelines that are tied to that. And so again, working with our nephrology colleagues—Dr. Agarwal and Dr. Wish—very closely to define the scope of practice of that pharmacist.
Things like credentialing requirements—so there’s privileging and credentialing that’s related to this. Some CPAs may have requirements in terms of the degree that’s required, maybe a residency, as well as in some cases, demonstrated competencies that might involve working with the physician to walk through some case examples and things just to ensure that there’s competency in the care of those patients under those disease states.
Slide 67

Different components of the CPA—again these are taken right out of CPAs that are for anemia management—such as initiating and evaluating patients on ESA therapy, initiating and evaluating patients based on iron deficiency anemia, and helping the care in terms of the guidelines around iron deficiency.
Ordering and evaluating lab results that correlate with the care and scope of practice there. And things that we’ve already talked about with looking at absolute and functional iron deficiency and things like that—things that need to be ruled out—or looking at B12 and folate in terms of macrocytic anemias that need to be ruled out before the ESA is instituted.
And as an agent of the physician, the prescriptions that are related to anemia management—things like ESA and iron—then can be transmitted. Those prescriptions can be transmitted and sometimes spelled out exactly how that can happen. And then, certainly, documentation of all that activity within the electronic medical record (EMR) or electronic health record (EHR) at the institution.
Slide 68

The dosing that’s typically followed is through the 2012 KDIGO guidelines published in Kidney International as a supplement and highlighted as the reference in this slide. There are also a number of parts of that that really highlight the increasing need for the care of anemia management in CKD and different GFR categories, as was discussed earlier. The prevalence of anemia increases as GFR drops, and really, you can see the nondialysis-dependent patients with CKD, really constitute about 50% of the anemia that we see in that population.
Slide 69

The CPA, again, is signed by the practitioner, whether the pharmacist in that particular clinic, as well as often the medical director. And these are fictitious names that I made up here, but these are some of the positions of different groups that would be involved in signing the CPA with the medical director of the CKD program. Sometimes the ambulatory care pharmacy manager within your system would be involved in endorsing this, as well as the director of pharmacy as a champion for this type of practice.
Slide 70
For the last part of my talk, I’ve included some of the CPAs—just some different examples of larger health systems, like major academic health centers, some of the regional and metro health centers, as well as the managed care system.
Slide 71
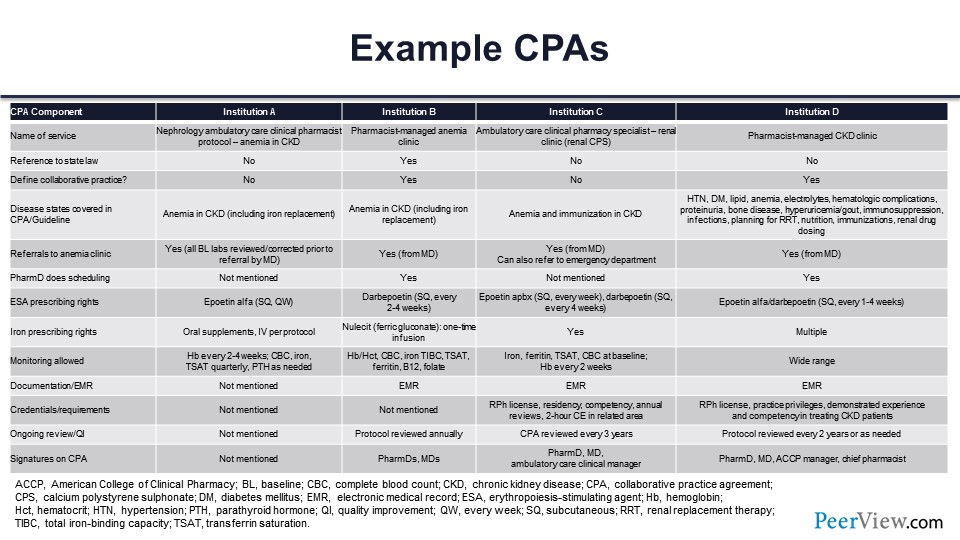
And I provided kind of a summary table that you can review that really outlines some of those key components and some of the CPAs that include different aspects, such as the name of the service—in some cases, it’s a nephrology ambulatory care clinical pharmacist within the CKD clinic. It could be a pharmacist-managed anemia clinic. All the way out to the far right-hand side is a real broad scope CKD clinic that really is just a pharmacist-managed CKD clinic that really sees a number of different disease states.
Often the state laws are referenced within the CPA. The CPA is defined in some cases. The different disease states are highlighted and on the left-hand side—anemia specifically in CKD, all the way out to more general and multiple disease states. Referrals to the anemia clinic, in all cases, need to take place. So the physician is referring that patient who’s diagnosed with CKD with anemia to that clinic. In some cases, there can be referral from the PharmD then out to things like the emergency department if something acute is happening, so there is a scope of practice around that as well.
Certainly, the prescribing rates of ESA are preapproved but can range to all the different ESAs. It also includes iron prescribing rates, different types of monitoring that’s allowed, documentation in the EMR, and in most cases, the credentialing, as well as ongoing review. The CPAs are typically reviewed on either an annual or biannual basis by the physician who’s overseeing that and the pharmacist. And then, lastly, the signatures on the CPA. So these are just some examples of four different institutions and kind of the broad array of different ranges of scope of practice within those.
Slide 72

So I think the last couple of things here are some case examples that we can work through. Let’s take just take a couple of minutes to look through, or walk through, some of these cases here.
This case is a 54-year-old man with diabetes, hypertension, and coronary artery disease. He’s being treated for CKD. He has an eGFR that’s been declining over the past 2 years. You can see here almost a 50% drop in GFR. The patient reports increased fatigue, and he asks about the causes of his anemia. So patients typically are not aware that they have CKD, and they’re not aware of anemia being present as well in CKD. In this case, his hemoglobin is 8.5 g/dL. TSAT is 20%, and ferritin is 320 ng/mL. The serum folate and vitamin B12 are found to be normal.
What do you think in this case is the most likely cause of this patient’s anemia? Would that be diabetes, relative erythropoietin deficiency, iron deficiency, multiple myeloma, or you’re not sure? Relative erythropoietin deficiency—so that’s the most common cause of anemia in CKD patients. In this case, the iron, TSAT, and ferritin were within the normal range.
Slide 73

Okay, so let’s go ahead and take a look at the second case. Here, we have a 76-year-old woman with diabetes and stage 4 CKD being evaluated for progression of anemia. Blood test results are notable for the serum potassium of 5.5 mEq/L and serum creatinine of 3.2 mg/dL. Serum calcium is at 12.6 mg/dL. Her hemoglobin is 7.5 g/dL, with normal erythrocyte indices. The serum ferritin is 358 ng/mL, and her TSAT is 20%.
In addition to erythropoietin deficiency, again she’s got stage 4 CKD, what other cause of anemia is important to exclude in this case, or what might need to be worked up further in this particular patient? So would that be iron deficiency, hyperparathyroidism, a malignancy, hypothyroidism, endocarditis, or other? So in this case, what we’re seeing is a patient with an elevated serum calcium, which often indicates a malignancy of some kind, and so that would be the case. I didn’t know if Dr. Agarwal or Dr. Wish would like to comment on some patients—maybe what you’ve seen with this type of scenario.
Dr. Wish: Absolutely, but we’d also consider hyperparathyroidism.
Dr. Dowling: As well, yes.
Dr. Wish: It can cause anemia because it disrupts the bone marrow.
Dr. Agarwal: I agree.
Slide 74

Dr. Dowling: Okay, and the last case, as a clinical pharmacist in the multidisciplinary CKD clinic, you’re managing patients with anemia under a CPA. In this case, you have a 50-year-old woman with stage 4 CKD present to your clinic for a follow-up. Two months ago, she was started on ESA, darbepoetin, 60 mcg subcutaneous every 2 weeks for her anemia, and that resulted in hemoglobin of 9.4 g/dL. Two weeks ago, the dose of that ESA was increased to 100 mcg subcutaneous every 2 weeks. Today, her hemoglobin is 11.2 g/dL; so in a 2-week period, she’s had an increase of almost 2 g/dL. Her ferritin is currently 300 ng/mL, and her TSAT is at 20%.
So given this response to that ESA therapy, what would you do in terms of a next step in this patient’s anemia management? Would you continue the ESA at the current dose and recheck hemoglobin in a month? Would you reduce the ESA dose to 80 mcg subcutaneous every 2 weeks and then recheck hemoglobin in a week? Would you hold the darbepoetin and recheck hemoglobin in 2 weeks? Would you administer some iron, like 1,000 mg IV iron here? Or would you discontinue the ESA therapy? Looking at that rapid rise in hemoglobin, the patient is still needing ESA therapy, but more than a 2 g/dL increase per month would institute a hold of the ESA and then a recheck within 2 weeks.
Dr. Wish: This is where the label disagrees with the KDIGO guidelines. The KDIGO guidelines actually discourage holding the ESA and reducing the dose, whereas the FDA has found it more appropriate to hold the dose.
Slide 75

Dr. Dowling: So in summary, again the role of the pharmacist in these emerging therapies, we’re going to see an increasing role of pharmacists. We have a number of studies that have shown the benefits of pharmacist-managed CKD clinics. That’s well documented in the peer-reviewed literature. There is expanding CPAs that are going to include potentially the new, emerging therapies; as they come on the market, they will be incorporated into these algorithms. And pharmacists will be working closely our physician colleagues on monitoring those patients, specifically for things like the cardiovascular outcomes and things like Dr. Wish described in terms of the potential adverse events. And then advanced practice pharmacists, really, it’s a great time if you’re a trainee, a resident, or a student. It’s a great time to be involved in ambulatory care pharmacy and anemia management clinics. There will be an increasing role going forward in these value-based care settings as well.
Audience Q&A
Slide 76

Dr. Wish: I can see we have lots of great questions. The first is, “Is your institution or healthcare organization treating the biosimilars as equivalent to the innovator products in regard to label versus unlabeled indications and dosing?” My institution is. Anil and Tom?
Dr. Agarwal: No.
Dr. Dowling: We’re seeing a movement to the biosimilars.
Dr. Wish: Yes, they’re moving to the biosimilars because they’re less, and there are really no signals that they’re any less effective or any less safe than the originator products.
Slide 77

Dr. Wish: “For patients with ESRD on hemodialysis or peritoneal dialysis, do you apply different levels to initiate iron replacement therapy and to evaluate iron and study ferritin and iron saturation?” The KDIGO guidelines suggest trying to achieve a ferritin level of at least 200 ng/mL in patients on hemodialysis, and 100 ng/mL in patients on peritoneal dialysis and patients not on dialysis because of the ongoing iron losses in the former.
Slide 78

Dr. Wish: “What do you recommend as the maximum epoetin and darbepoetin dose? Do you also use weight-based dosing, or do you have a max single dose that you primarily do not exceed?”
And the answer is we actually use both, and it varies from nephrologist to nephrologist. My personal maximum, which is pretty much the policy in my dialysis unit, is the equivalent of 100 U/kg body weight three times weekly for epoetin, so that would be about—if it’s a 70-kg person—about 7,000 or somewhere in the neighborhood of 20,000 U/week. In darbepoetin dosing, that would be approximately 100 U/week. Same?
Dr. Agarwal: Same.
Dr. Dowling: Same.
Slide 79

Dr. Wish: Okay, great. “What is the clinical significance of differences between HIF-PHD inhibitors in terms of HIF stabilization and enzyme isoform inhibition?”
And the answer is, we don’t know. Until there are head-to-head studies between these agents, we cannot tell whether some of these theoretical specificities of PH 2, PHD 2 versus PHD 3, and more potentiation of HIF-2-α versus HIF-1-α has any significance in terms of clinical practice. So my answer is we don’t know; we need more data. Anybody want to add to that?
Dr. Agarwal: Exactly.
Slide 80

Dr. Wish: “Can the published roxadustat data in Chinese patients be extrapolated to other patient populations?” And the answer is we’re not really sure. I mean, the Chinese patients are a little different. And remember, the Chinese studies, the phase 3 were a maximum of 6 months, so we don’t have any kind of long-term safety signals with regard to MACE.
We know that they were effective in patients with and without inflammation who were on hemodialysis. They were in patients with inflammation and CKD. There was the issue with hyperkalemia, which interestingly was noted in the Chinese population and has not been mentioned in any of the other worldwide studies. So the answer is extrapolation, I think, to a certain extent is limited.
Dr. Agarwal: Yes.
Slide 81

Dr. Wish: “What are the primary considerations for pharmacists in the use of HIF-PH inhibitors—selecting among the three? Adverse events?” And the answer is, you know, I personally see the pharmacist role with the HIF-PH inhibitors to be the same as we have with ESAs. I don’t think that’s going to change the paradigm at all.
Right now, the selection of the agent, once all three are approved by the FDA, is inevitably going to be based on the value proposition. What is the price? And I anticipate, since many of these in the non-CKD population will have to undergo a process of prior approval, the preferred agent will probably be the one that’s least expensive. I don’t see any significant efficacy or safety differences between the three agents, especially in the absence of head-to-head studies, so I think it will all boil down to price.
Dr. Dowling: Yeah, I agree, and I think with talking to the patients, since these are orally available, especially in the nondialysis-dependent population, this will be an option for them to talk through in terms of moving from subcutaneous to an orally available agent. If they feel more comfortable with that, that’s going to be part of the factor as well I think.
Dr. Wish: Well, and I agree, and I think one of my concerns, and it may be shared by other panel members, is that the payers are just going to look at the price. They’re not going to look at any other patient-friendly or patient-specific considerations, and they may require that a patient fail ESA therapy before they’ll approve a HIF stabilizer in the nondialysis-dependent population. In the dialysis population, these decisions will be made by the dialysis companies. You know, these are going to be included in the bundle eventually, so the dialysis providers will get a lump sum of money, and then they’ll have to decide how they want to spend it with regard to who gets ESAs and who gets HIF stabilizers.
Slide 82

Dr. Wish: “Do either Dr. Wish or Dr. Agarwal work with pharmacists in their clinics? And if so, how do they utilize them as members of the care team?”
So my answer is, I do not have a pharmacist in my CKD clinic. I did have a pharmacist in my dialysis unit when my dialysis unit was owned and operated by my academic medical center, and that pharmacist was a full member of the team. She made rounds with us on all the patients all time. She was absolutely indispensable in terms of pointing out drug-to-drug interactions, working with the patients directly to make sure that they filled their prescriptions, and ensuring that we did drug dose reconciliation, if there were any changes in hospitalizations, etc. She would also point out patient assistance programs that we would not be aware of. So I mean, she was worth her weight in gold. And then what happened is that our dialysis unit was sold to one of the large chains not to be mentioned, and the pharmacist was eliminated. And I miss her.
Dr. Agarwal: We do have a pharmacist, and they assist with everything that you said, plus also in regulating tightly the dosages of ESA with us, reporting the hemoglobin levels. But we do it in combination; they don’t have a CPA like you were talking about, but they will independently do it. But we do work closely with them.
Slide 83

Dr. Wish: “Can you describe the monitoring needed with HIF-PHIs? Do you see the role of the pharmacist changing with introducing HIF?” And the answer is no. I see the role as being the same. I think the need to monitor for the rate of rise of hemoglobin or overshoot of hemoglobin may be less with the HIFs because we’re not seeing the same safety signals that we saw with ESAs.
And I think it all boiled down to the label. I mean, if the label has similar black box warnings as the ESAs, which the FDA may or may not decide to do, then the role of the pharmacist will be identical in terms of monitoring for the rate of rise of hemoglobin, the peak hemoglobin level. If the FDA decides that the safety of the HIF stabilizers is greater or better than with the ESAs, and we don’t have all these black box concerns in terms of the rate of rise, then I think that will make the pharmacist’s job a lot easier data.
Dr. Agarwal: Right, I totally agree. So, because these studies were all short term, and we need longer-term studies and the real-life experience probably, which will tell us whether or not these are safe or if these are safer. Maybe we could revisit the target hemoglobin levels again because previously all the studies were with the ESA as that was the only agent, and higher targets were not good. But maybe if it was the ESA effect and not the hemoglobin target effect, then it might be easier to use these agents and get the hemoglobin to a higher level, and patients might have better QOL. I don’t know. It’s to be seen.
Dr. Wish: Right. So that’s my last question and answer. Certainly, I’ like to thank our speakers, Dr. Dowling and Dr. Agarwal, for a couple of really outstanding presentations.
Back Top