
Expired activity
Please go to the PowerPak
homepage and select a course.
New Directions for Oral Treatment in B-Cell Malignancies: Implications for Pharmacists
INTRODUCTION
Non-Hodgkin lymphoma (NHL) is a heterogeneous group of B- and T-cell malignancies ranging from indolent to very aggressive subtypes.1 The most common aggressive NHL is diffuse large B-cell lymphoma, while follicular lymphoma (FL) and chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) are the most common indolent lymphomas. Waldenström macroglobulinemia (WM) is a rare but important subtype of NHL that is also characterized by an indolent disease course. Mantle cell lymphoma (MCL) is a heterogeneous subtype of NHL that can present with features of both indolent and aggressive NHL. In the United States (U.S.), there will be an estimated 77,240 new cases of NHL and 19,940 deaths related to these malignancies in 2020. SLL, but not CLL, is incorporated into the aforementioned NHL statistics. Of CLL alone, there will be an additional 21,040 new cases and 4060 deaths. Cumulatively, NHL ranks among the top 10 leading cancer types in the U.S. for new cases and deaths for both males and females.2
For decades, cytotoxic chemotherapy and chemoimmunotherapy have been the primary treatment modalities for patients with NHL. Unfortunately, a large portion of patients will not be cured and are subjected to additional lines of therapy along with acute and long-term treatment-related adverse effects.1 Considering that the median age at diagnosis of NHL is approximately 67 years of age, intensive chemotherapy options are not an attractive treatment approach due to toxicities and poor tolerability.1
In an attempt to reduce treatment-related complications and improve disease control, novel therapies have been and continue to be incorporated into the treatment paradigm of NHL. Agents targeting the B-cell receptor (BCR) pathway such as Bruton’s tyrosine kinase (BTK) inhibitors (ibrutinib, acalabrutinib, and zanubrutinib), phosphatidylinositol-3-kinase (PI3K) inhibitors (idelalisib, duvelisib, and copanlisib), and an agent targeting the BCL2 pathway (venetoclax) have changed treatment paradigms across various subtypes of NHL.
PATHOPHYSIOLOGY AND MECHANISMS OF ACTION
The BCR pathway regulates B-cell differentiation, proliferation, and chemotaxis. Antigen and/or ligand-independent signaling through the BCR leads to abnormal intracellular signal transduction of malignant B-cells.3,4 Several signaling targets and pathways such as LYN, spleen tyrosine kinase (SYK), BTK, phospholipase-Cγ2 (PLCγ2), protein kinase C (PKC), PI3K, nuclear factor (NF)-kβ, MAPK/ERK, and others may be aberrantly activated.5,6 Overexpression of these targets and/or pathways plays an essential role in CLL, MCL, FL, and WM pathogenesis. BTK and PI3K are common convergent points and, therefore, are attractive targets to halt excessive signaling through the BCR. As a result, inhibition of BCR can abrogate many cellular processes via BTK or PI3K. These include but are not limited to B-cell selection, differentiation, proliferation, motility, homing, adhesion, chemotaxis, and survival (Figure 1).5,6
Ibrutinib, acalabrutinib, and zanubrutinib covalently bind to the cysteine 481 residue of BTK, leading to irreversible inhibition of BTK. However, ibrutinib also inhibits several “off-target” enzymes such as TEC, ITK, BMX, EGFR, ERBB4, JAK3, BLK, and others. Acalabrutinib and zanubrutinib were developed to have less or no inhibition of these enzymes with the goal of ameliorating off-target toxicity.7
The PI3K inhibitors (idelalisib, duvelisib, and copanlisib) also have differences based on their various binding affinities. PI3Ks are cytoplasmic tyrosine kinases that can be divided into 4 major catalytic isoforms–alpha (α), beta (β), gamma (γ), and delta (δ)–and 3 classes–I, II, and III. Class I PI3Ks have been studied most extensively and comprise the 110-kDa catalytic subunits of p110: α, β, γ, and δ. The p110-δ isoform is primarily expressed in B and T lymphocytes, the γ isoform is expressed in granulocytes and T lymphocytes, the β isoform is expressed within platelets, and the α isoform is primarily responsible for glucose homeostasis. Idelalisib predominantly inhibits δ, duvelisib inhibits δ and γ, and copanlisib inhibits all 4 isoforms.8–14 The differential selectivity for each isoform among the 3 PI3K inhibitors predicts their toxicity profiles.
The BCL2 pathway is a separate independent pathway with the principal function of controlling apoptosis. This pathway consists of 3 subclasses: the promoters of cell death collectively known as BH3 proteins (BIM, BAD, PUMA, and NOXA), pro-survival proteins (BCL2, BCLxL, MCL1, and BFL1/A1), and mediators of cell death (BAX and BAK).15 Various cellular stimuli activate the BCL2 pathway, which mobilizes the BH3 proteins. Typically, the pro-survival proteins are bound to and inhibit the function of the cell death mediators (BAX and BAK). When the BH3 proteins bind to BCL2 (or other pro-survival proteins), BAX and BAK are released from BCL2.16 The release of BAX and BAK causes the mitochondrion to increase its outer membrane permeability, destroying the mitochondrion and the efflux of cytochrome c. This process activates caspases, which degrade the cell.17,18 Malignant cells utilize this pathway for a survival advantage by overexpressing pro-survival proteins such as BCL2.19 With an abundance of BCL2, the BH3 proteins are overwhelmed, the cell death mediators (BAX and BAK) cannot be released and cell death cannot occur. Venetoclax acts as a BH3 protein mimetic (or a BCL2 antagonist) by binding to BCL2 and releasing BAX and BAK, potentiating cellular apoptosis.
Figure 1. The BCR and BCL2 Pathway5,6
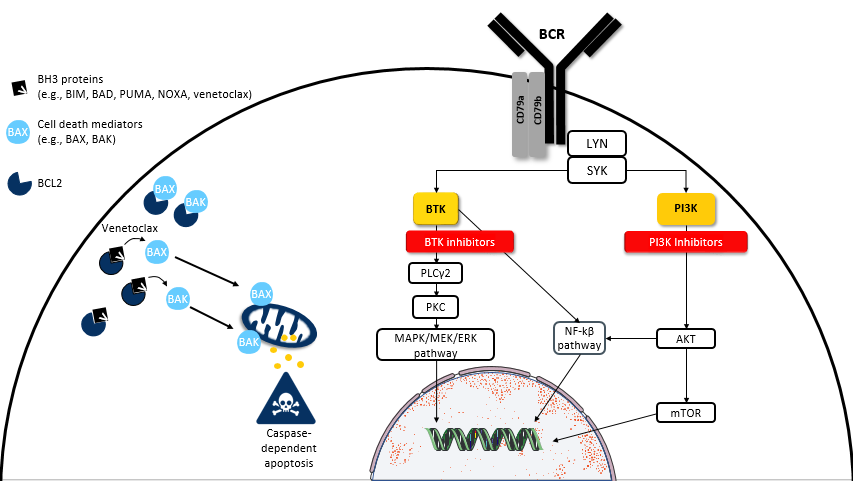
BCR, B-cell receptor; BTK, Bruton’s tyrosine kinase; NF-kβ; nuclear factor-kβ; PI3K, phosphatidylinositol-3-kinase; PLCγ2, phospholipase-Cγ2; PKC, protein kinase C; SYK, spleen tyrosine kinase.
Disease state presentation and treatment
Chronic lymphocytic leukemia/small lymphocytic lymphoma
CLL is diagnosed when a patient has > 5000 clonal lymphocytes in their blood. SLL is characterized by the presence of lymphadenopathy and/or splenomegaly composed of cells phenotypically identical to CLL but patients have < 5000 clonal lymphocytes in the blood.20 CLL and SLL are considered to have the same cell of origin (but with differing presenting features), so their treatment approach is identical. Median age at diagnosis is 70 years and many patients will have significant comorbidities that will preclude them from intensive therapy.21
There are several disease-related prognostic features that have been reported for CLL/SLL: del(17p)/TP53 mutation, immunoglobulin heavy chain (IGHV) status, and a complex karyotype (3 or more chromosomal abnormalities) are arguably the most relevant.20 Patients with a mutation in TP53, del(17p), a complex karyotype, and unmutated IGHV have poor and short-lived responses to traditional chemotherapy.22 Conversely, patients with mutated IGHV are exquisitely responsive to chemotherapy and may have a chance for cure or the potential for a durable remission with fludarabine, cyclophosphamide, and rituximab (FCR).23–26
Patient case 1: NV is a 59-year-old male with newly diagnosed CLL (IGHV mutated, TP53WT) and complaining of significant fatigue, early satiety, and night sweats that keep him up at night. He has no other significant past medical history and is presenting today to discuss first-line treatment options. Which of the following is the best therapy for NV?
- FCR
- Ibrutinib + rituximab
- Venetoclax + obinutuzumab
- Acalabrutinib
Prior to 2018, the treatment of CLL could be divided into 4 major categories according to patient age and status: patients who are young/fit (< 60-65 years), older/fit (> 60-65 years), and older/unfit (> 65 years) with comorbidities, and patients with del(17p) or a p53 mutation. Young/fit patients were treated with chemoimmunotherapy (FCR or bendamustine plus rituximab [BR]), older/fit patients were treated with BR, older/unfit patients with comorbidities were treated with obinutuzumab plus chlorambucil or ibrutinib, and patients with del(17p) or a p53 mutation were treated with ibrutinib. Indeed, significant overlap existed throughout the aforementioned groups, but, for simplicity, this categorization should serve as the benchmark for understanding the progression toward today’s treatment algorithm.
The ECOG 1912 trial was a landmark prospective, multi-center, open-label, randomized controlled phase 3 trial that changed practice for young/fit patients.27 Treatment-naïve patients £ 70 years old without significant comorbidities and an ECOG performance status of 0–2 were randomized 2:1 to receive ibrutinib 420 mg orally daily continuously with rituximab on cycles 2–7 (n = 354) or FCR for 6 cycles. Patients with del(17p) were excluded. At 3 years, the primary endpoint of progression-free survival (PFS) was improved with ibrutinib plus rituximab compared to FCR (3-year PFS: 89.4% vs. 72.9%, p < 0.001). PFS in individuals with unmutated IGHV demonstrated a dramatic benefit with ibrutinib plus rituximab (3-year PFS: 90.7% vs. 62.5%, p < 0.001). Longer follow-up is required to determine the impact of ibrutinib plus rituximab in patients with IGHV-mutated CLL. As a result of this trial, the National Comprehensive Cancer Network (NCCN) CLL guidelines recommend ibrutinib with or without rituximab as a first-line option for young/fit patients (Table 1). Patients with IGHV-mutated CLL without del(17p) can still be considered for FCR, but ibrutinib with rituximab is also an NCCN-recommended option for these patients.20
| Table 1: Summary of Novel Agents Used in the Treatment of Lymphoma 20,32,35-39,42,46,57,59,61 |
Agent |
FDA approved indication (line of therapy) |
Class |
Dose and route |
How supplied |
| Ibrutinib |
CLL (1st+), MCL (2nd+), WM (1st+) |
BTK inhibitor |
MCL: 560 mg orally daily
CLL: 420 mg orally daily |
70-, 140-, 280-, 420-, and 560-mg tablets |
| Acalabrutinib |
MCL (2nd+), CLL (1st+) |
BTK inhibitor |
100 mg orally twice daily |
100-mg capsule |
| Zanubrutinib |
MCL (2nd+) |
BTK inhibitor |
160 mg orally twice daily or 320 mg orally daily |
80-mg capsule |
| Idelalisib |
CLL (3rd+),
FL (3rd+),
CLL with comorbidities (2nd+) |
PI3K inhibitor (δ) |
150 mg orally twice daily |
100- and 150-mg tablets |
| Duvelisib |
CLL (3rd+),
FL (3rd+) |
PI3K inhibitor (γ/δ) |
25 mg orally twice daily |
15- and 25-mg
capsules |
| Copanlisib |
FL (3rd+) |
PI3K inhibitor (α/δ) |
60 mg IV over 1 hour days 1, 8, and 15 (28-day cycle) |
60-mg vial |
| Venetoclax |
CLL (1st+) |
BCL2 inhibitor |
MCL*: 400–800 mg orally daily with food
CLL: 400 mg orally daily with food |
10-, 50-, and 100-mg tablets |
BTK, Bruton’s tyrosine kinase; CLL, chronic lymphocytic leukemia; FDA, United States Food and Drug Administration; FL, follicular lymphoma; IV, intravenously; MCL, mantle cell lymphoma; PI3K, phosphatidylinositol-3-kinase; WM, Waldenström macroglobulinemia.
*Not FDA approved for MCL. |
The ALLIANCE trial was another cooperative group study that led to a practice change in CLL.28 This was a prospective, multi-center, open-label, phase 3 clinical trial that randomized older/fit (³ 65 years) patients with an ECOG performance status of 0–2 and without significant comorbidities 1:1:1 to ibrutinib 420 mg orally daily continuously with rituximab, ibrutinib (same dose) alone, or BR for 6 cycles. Patients with del(17p) were excluded. At 2 years, the primary endpoint of PFS was significantly improved in both arms receiving ibrutinib compared to BR (2-year PFS: 87% vs. 88% vs. 74% for ibrutinib with rituximab, ibrutinib alone, and BR, respectively, p < 0.001). Two major practice changes occurred as a result of this study. First, BR is no longer the standard of care for older/fit patients with CLL. Second, since there was no improvement in PFS with the addition of rituximab to ibrutinib, single-agent ibrutinib without rituximab is an appropriate option for first-line CLL treatment. Taking the ECOG 1912 and ALLIANCE trials together, it is now appropriate for patients to receive a chemotherapy-free approach in the front-line treatment setting for CLL.
Chemotherapy-free treatment is also available for older/unfit patients and has been a standard for patients with del(17p). The CLL14 trial added venetoclax with obinutuzumab to the treatment arsenal for older/unfit patients.29 This trial was a prospective, multi-center, open-label, phase 3 study that randomized patients 1:1 to venetoclax 400 mg orally daily (following a weekly dose escalation that began on day 22 of cycle 1) for a total of 12 cycles with obinutuzumab for 6 cycles versus chlorambucil (12 cycles) with obinutuzumab (6 cycles). Patients were treatment naïve but had significant comorbidities (a total score of > 6 on the Cumulative Illness Rating Scale) or creatinine clearance < 70 mL/min. Patients with del(17p) were also included, which is in contrast to the ECOG 1912 and ALLIANCE trials.27,28 The primary endpoint, PFS, was improved in the venetoclax arm compared to the chlorambucil arm (2-year PFS: 88% vs. 64%, p < 0.001). Expectedly, the benefit was also seen in patients with del(17p). On the basis of these results, venetoclax with obinutuzumab is now a standard-of-care option for older/unfit patients or patients with significant comorbidities (Table 1). It is placed at a similar recommendation level by NCCN with ibrutinib for this population and for patients with del(17p), but it allows for a finite duration of therapy (1 year), whereas ibrutinib is continued until progression (often several years to possibly greater than a decade).20
Despite the success of ibrutinib, there have also been challenges. Ibrutinib may be difficult to tolerate for select patients due to both its on-target (BTK) and significant off-target (TEC, ITK, EGFR, JAK3, BLK, etc.) effects. As a result, researchers have sought to identify alternative BTK inhibitors with fewer off-target effects to improve the tolerability profile for patients. One such agent is acalabrutinib. The ELEVATE-TN trial evaluated a similar population to the CLL14 trial with a cohort consisting of older/unfit patients or those with significant comorbidities who were treatment naïve. This study was a prospective, multi-center, open-label, phase 3 trial that randomized patients 1:1:1 to acalabrutinib 100 mg orally every 12 hours continuously with obinutuzumab (cycles 2–6), acalabrutinib 100 mg orally every 12 hours continuously as monotherapy, or obinutuzumab with chorambucil.30 At 30 months, the primary endpoint of PFS was significantly improved in both arms receiving acalabrutinib compared to chlorambucil with obinutuzumab (30-month PFS: 90% vs. 82% vs. 34% for acalabrutinib with obinutuzumab, acalabrutinib alone, and chlorambucil with obinutuzumab, respectively, p < 0.0001). When comparing the 2 acalabrutinib arms, there is currently no significant difference in PFS; however, there is a higher response rate with adding obinutuzumab to acalabrutinib (94% vs. 79%, p < 0.0001). As such, acalabrutinib +/– obinutuzumab is recommended alongside ibrutinib and venetoclax with obinutuzumab as a front-line treatment option for older/unfit patients with CLL (Table 1).20 Further studies are required to elucidate the role of obinutuzumab when combined with oral targeted therapies and to determine the optimal sequence and combinations of oral targeted therapies in CLL.
Patient case 1 (continued): Initial and subsequent therapy selection depends on patient age, comorbidities, and patient and provider preferences. In patient NV, FCR, ibrutinib +/– rituximab, venetoclax + obinutuzumab, and acalabrutinib are all appropriate options: in a patient such as NV, who has no comorbidities, the choice truly comes down to preference. A convincing argument can be made to treat NV with FCR on the basis of data suggesting the potential for long, durable remissions and/or cure for patients with IGHV-mutated CLL. FCR also provides a finite (6 cycles) duration of therapy. On the other hand, BTK inhibitors and venetoclax-based regimens may also provide long, durable remissions without the short- and long-term risks of cytotoxic chemotherapy; however, longer follow-up is required to elucidate this potential. Additionally, it remains unknown whether adding rituximab/obinutuzumab to targeted therapies can improve long-term outcomes.
Mantle cell lymphoma
MCL is an interesting and unique subcategory of B‐cell NHL with a generally aggressive, albeit heterogeneous, clinical course. MCL accounts for about 3%–10% of adult‐onset NHL in western countries and shows historically poor long-term survival compared to other B-cell malignancies.1,31 Recent progress in the understanding of the biology of MCL has led to improvements in patient outcomes and the use of several novel targeted therapies.
MCL is classically defined by the t(11;14)(q13;q32) translocation, which juxtaposes the CCND1 gene encoding cyclin D1 to the IGHV, resulting in the overexpression of cyclin D1.31 Alternatively, less-frequent alterations in CCND2 and CCND3, encoding cyclin D2 and D3, respectively, have been identified in MCL lacking the classical t(11;14)(q13;q32) translocation. Additional markers found in MCL include SOX11 overexpression, hypermutated IGHV, and TP53 mutations. While the prognostic impact of SOX11 is unknown, patients with TP53 mutations or with complex karyotypes generally have a poor prognosis. On the other hand, patients with IGHV mutations typically show better outcomes.
Patients with MCL have varied clinical presentations,31 ranging from asymptomatic lymphocytosis or non‐bulky nodal/extranodal disease with minimal symptoms to progressive generalized lymphadenopathy, cytopenias, splenomegaly, extranodal disease (including involvement of various sections of the gastrointestinal tract, the kidneys, the central nervous system [although rarely], or any other organ system) with significant symptom burden. The majority of patients (70%–80%) will present symptomatically and require some form of systemic therapy.
The treatment approach can vary and depends on symptom burden and patient fitness. Initial treatment for younger (< 65 years of age), fit patients commonly comprises 2 components: cytarabine-containing intensive induction chemotherapy followed by an autologous stem cell transplant (ASCT).32 For those who are older and/or unfit, intensive therapies, including ASCT, are not an option. In this scenario, clinical judgement of patient status and comorbidities should guide the selection of a less-intensive chemotherapy plan, of which several suitable options exist (e.g., bendamustine-based regimens).32 Both fit and unfit patients are recommended to receive rituximab maintenance for 3 years after first remission due to a significant improvement in PFS.32,33
MCL is considered an incurable NHL and, thus, the vast majority of patients will eventually relapse. Relapsed MCL has been historically characterized by poor response rates and overall survival (OS) of < 3 years.34 There are numerous options for second or later relapse for MCL, including chemoimmunotherapy and several chemotherapy-free approaches.32 Therapy selection for relapsed or refractory (R/R) MCL should follow similar logic as other NHLs, where the responses and durations of response to previous therapies are examined along with patient age, performance status, and comorbidities.
For those with a prolonged response to first-line therapy (> 24 months), chemoimmunotherapy remains a suitable option, as these patients may still show ‘chemosensitive’ disease. For those who relapse early (< 24 months), a more novel approach is appropriate, as these patients are felt to be more ‘chemoresistant’ and likely harboring a TP53 mutation. In this setting, the development of BTK inhibitors (ibrutinib, acalabrutinib, and zanubrutinib) and a BCL2 antagonist (venetoclax) has revolutionized treatment options for R/R MCL (Table 1).
Ibrutinib is the most extensively studied BTK inhibitor for R/R MCL. Three main trials have been conducted to date: PCYC-1104 (phase 2), which led to the approval of ibrutinib in MCL, SPARK (phase 2), and RAY (phase 3). All 3 were pooled into 1 analysis that included 370 total patients.35 Patients received ibrutinib 560 mg orally once daily continuously. Of the included patients, 73% had received ≥ 2 lines of prior therapy (median, 2; range, 1–9) and 12% had blastoid variant histology (a more aggressive variant of MCL). Overall response rate (ORR) in the entire population was 70%, with a 27% complete remission (CR) rate. Median PFS and OS were 12.5 and 26.7 months, respectively. Patients receiving ibrutinib in the second line had improved outcomes compared to those treated in later lines. Median PFS in patients who received 1 line of prior therapy was 25.4 months with an OS that has not yet been reached; in those with > 1 line of prior therapy, the median PFS was 10.3 months and OS was 22.5 months. Of 144 patients with known TP53 mutation status, 20 (14%) had mutated TP53. In patients with mutated and wild-type TP53, median PFS was 4 and 12 months and median OS was 10.3 and 33.6 months, respectively; ORR in patients with mutated and wild-type TP53 was 55% (11 partial responses [PRs], 0 CRs) and 70% (56 PRs, 31 CRs), respectively.
Acalabrutinib was approved for R/R MCL on the basis of the phase 2, single arm, multicenter ACE-LY-004 trial, which included patients with R/R MCL who had received 1–5 prior lines of therapy.36 To be eligible, patients could not have received a prior BTK inhibitor or BCL2 antagonist, have notable cardiovascular disease (class 3/4 cardiac disease per New York Heart Association Functional Classification, congestive heart failure, or myocardial infarction [MI] within 6 months of screening, uncontrolled or symptomatic arrhythmias, or QTc > 480 ms), or be on a concurrent vitamin K antagonist. Patients received acalabrutinib 100 mg orally twice daily continuously. Of 124 included patients, the median number of prior therapies was 2 (range, 1–2). Acalabrutinib demonstrated an ORR of 81% with a CR rate of 43%. The median PFS was 20 months and estimated 24-month OS was 72%, as median OS had not yet been reached. TP53 mutational status was not explored in this analysis.37
Zanubrutinib was studied for MCL in the phase 2 single-arm BCG-3111-206 trial that included 86 patients with 1–4 prior lines of therapy (no prior BTK inhibitors) and no notable cardiovascular disease.38 The median number of prior therapies was 2 (range, 1–4) and 34% of patients had at least 3 prior therapies. In all, 28% of patients included had TP53 mutations. Zanubrutinib 160 mg orally twice daily was given continuously. The ORR of 84% was similar to that of other BTK inhibitors. While the CR rate of 69% was higher than previously reported with ibrutinib or acalabrutinib, comparisons across these single-arm BTK inhibitor studies are limited due to varying baseline patient populations. Median PFS was also similar at 22.1 months. Interestingly, patients with TP53-mutated tumors had an ORR comparable to those with wild-type disease (80.0% vs. 87.2%). Patients with TP53 mutations demonstrated decreased duration of response (14.5 months vs. 19.5 months) and PFS (14.7 months vs. 22.1 months) compared to patients without TP53-mutated tumors.
As a result of these analyses, all 3 BTK inhibitors are approved and appropriate for use in the R/R MCL setting.32 Efficacy may be improved if BTK inhibitors are used earlier on in the history of a patient’s disease course. Choice of 1 BTK inhibitor over another is difficult based purely on efficacy as cross-trial comparison is extremely limited. It seems, at this time, that the available BTK inhibitors demonstrate comparable efficacy in this setting. Currently, the choice of 1 BTK inhibitor over another is driven by differences in their toxicity profiles in the current state.
Venetoclax, while not approved by the U.S. Food and Drug Administration (FDA), is recommended in the NCCN guidelines as an option for R/R MCL either as a single agent or in combination with ibrutinib.32 Single-agent venetoclax (dose escalation to target dose of 200–1200 mg) was studied in the phase 1 M12-175 trial of 28 R/R MCL patients who received 1–7 prior lines of therapy (median, 3).39 Of note, no patients received prior BTK inhibitor therapy or lenalidomide. As this was a phase 1 trial with the primary outcome of safety, efficacy outcomes were limited. The ORR was 75% with a CR rate of 21%. The median time to response was 36.5 days and median estimated PFS was 14 months. Unfortunately, as no patients had received a prior BTK inhibitor in this analysis, the optimal place in therapy for venetoclax and the optimal sequence of BTK inhibitors and venetoclax are unknown. For patients with contraindications to BTK inhibitors or who may not tolerate BTK inhibitors, venetoclax remains a good option. Real-world evidence suggests that venetoclax does not retain significant activity after BTK inhibitor exposure, with an ORR of 53% (18% CR) and a median PFS of only 3.2 months.40 It is unclear if the reverse sequence (venetoclax prior to BTK inhibitor) impacts response to BTK inhibitor therapy.
Combination venetoclax and ibrutinib was studied in the phase 2 single-arm AIM trial.41 Twenty-four patients who had R/R MCL (n = 23) or were treatment naïve and not a candidate for chemoimmunotherapy (n = 1) were included. The number of median prior therapies was 2 (range, 1–6). No patients had prior BTK inhibitor or venetoclax exposure, although the protocol was amended to allow patients with prior BTK inhibitor exposure to enroll. Half (n = 12) of the patients had a TP53 deletion or mutation. Patients received single-agent ibrutinib 560 mg orally daily for 4 weeks prior to initiating venetoclax to reduce the risk of tumor lysis syndrome (TLS). Venetoclax (dose escalation weekly to 400 mg orally daily; increased to 800 mg orally daily if no CR by week 16) was introduced at week 5. ORR was 71% with a 62% CR rate. Median PFS was 29 months and median OS was 32 months. Of the patients who had a TP53 mutation or deletion, 50% had a CR. While the rationale for combining a BTK inhibitor with a BCL2 inhibitor is compelling, response rates demonstrated here do not appear to be dramatically different than those reported with single-agent BTK inhibitors. Differences in patient characteristics could account for this disappointing observation. At this time, the most appropriate patient for combination versus single-agent therapy is unknown. Furthermore, the trial did not enroll any patient with prior BTK inhibitor exposure; thus, it is unknown if combination therapy retains activity in these patients and, if so, if this retained activity is more robust than single-agent venetoclax. Nonetheless, combination ibrutinib and venetoclax in clinical practice may be reserved for patients with more aggressive disease activity or who are either slow to respond or who do not show optimal response (stable disease [SD] or PR) to single-agent ibrutinib.
More data are needed to guide the choice of 1 BTK inhibitor over another, to determine the optimal sequence of therapies in the R/R MCL setting, and to determine who, if anyone, is the most appropriate patient for combination BTK inhibitor and BCL2 antagonist. Nonetheless, all BTK inhibitors remain suitable options for patients with R/R MCL according to patient age, comorbidities, preference, safety profile, and cost.
Waldenström macroglobulinemia
WM is a rare, indolent subtype of B-cell NHL characterized by lymphoplasmacytic involvement of bone marrow or lymph nodes and the secretion of immunoglobulin M. Patients may present with a variety of clinical manifestations including hyperviscosity syndrome, neuropathy, adenopathy or organomegaly, amyloidosis, cryoglobulinemia, cold agglutinin disease, and/or cytopenias.42 Nearly 90% of patients will have a somatic mutation involving MYD88L265P, which activates the BCR pathway and can be inhibited downstream via BTK inhibition.43 A second important somatic activating mutation within the C-terminal of the C-X-X chemokine receptor type 4 (CXCR4) gene that bypasses BTK occurs in approximately 20%–40% of patients.43,44 As such, patients with mutations in CXCR4 (i.e., CXCR4WHIM) experience diminished benefit with BTK inhibitors.45
Patients who present with minor symptoms and minimal complications are diligently watched and treatment is withheld until significant symptoms arise. Patients with bulky disease, profound cytopenias, constitutional symptoms, or symptoms of hyperviscosity will prompt the need for treatment. An initial assessment for requiring plasmapheresis is made if hyperviscosity is present with significant associated symptoms. Following this assessment, many first-line therapy options are appropriate such as BR; ibrutinib +/– rituximab; dexamethasone, rituximab, and cyclophosphamide (DRC); and bortezomib, dexamethasone, and rituximab (BDR).
Similar principles are used to guide therapy selection in relapse. Patients who relapse late (> 3 years) can be treated with the original treatment regimen. Patients with R/R disease recurring earlier (< 3 years) can be treated with a BTK inhibitor (ibrutinib, acalabrutinib, or zanubrutinib), BDR, BR, or DRC, if not previously received (Table 1).42,46
The iNNOVATE trial was a prospective, multi-center, placebo-controlled phase 3 trial that randomized patients with WM who were not refractory to rituximab or had not received rituximab in the previous 12 months to ibrutinib 420 mg orally daily continuously until progression with rituximab or placebo with rituximab.45 Rituximab was administered weekly on day 1 of weeks 1–4 and 17–20 in both arms. At 30 months, the primary endpoint of PFS was improved with ibrutinib plus rituximab compared to rituximab alone (30-month PFS: 82% vs. 28%, p = not reported). The median PFS has not been reached for the ibrutinib arm, while the rituximab-alone arm demonstrated a median PFS of 20.3 months (p < 0.001). Of note, patients with a mutation in CXCR4 or without a mutation in MYD88 had lower and slower responses than those without CXCR4 or with an MYD88 mutation, respectively. As a result of this trial, the NCCN WM guidelines recommend ibrutinib as a first-line option with or without rituximab.42
The ASPEN trial was a prospective, multi-center, open-label phase 3 trial that randomized patients with WM who were either treatment naïve and not a candidate for chemoimmunotherapy or who had R/R WM.47 Patients were randomized 1:1 to receive either zanubrutinib (n = 102) 160 mg orally twice daily or ibrutinib (n = 99) 420 mg orally once daily. The primary endpoint of ORR was not different between the groups (94.1% vs. 92.9%, respectively). This was the first randomized controlled trial that compared 2 BTK inhibitors. Although there were no differences in ORR, PFS, or OS, zanubrutinib demonstrated an improved safety profile. Patients were less likely to stop zanubrutinib (4% discontinuation rate) than ibrutinib (9.2%). The incidences of atrial fibrillation (AF) (2% vs. 15.3%), hypertension (10.9% vs. 17.3%), and major bleeding (5.9% vs. 9.2%) were lower with zanubrutinib than with ibrutinib. However, zanubrutinib led to an increased incidence of neutropenia (29.7% vs. 13.3%).47 On the basis of these findings, one should expect to see an approval by the FDA and an NCCN guideline recommendation for the use of zanubrutinib for the treatment of WM in the future.
Acalabrutinib has also been studied in WM.48 The ACE-WM-001 trial was a phase 2, multi-center, open-label study evaluating acalabrutinib 100 mg orally daily for patients with mostly R/R WM. A small cohort of treatment-naïve patients who declined or had comorbidities that precluded treatment with chemoimmunotherapy were included. Acalabrutinib treatment resulted in an ORR of 93% in both treatment-naïve and R/R disease. As expected, patients with a mutation in MYD88L265P had a higher ORR (94%) than those with MYD88WT (79%). Acalabrutinib has not yet gained FDA approval for use in WM but could soon be a treatment option for patients with R/R WM.
It is important to note that, for all diseases discussed in this review, selecting a BTK inhibitor for a patient who has relapsed or is refractory following another BTK inhibitor would not be expected to show much additional activity; these 3 agents have overlapping mechanisms of resistance. Thus, if ibrutinib is used in front-line WM, ibrutinib, acalabrutinib, and zanubrutinib should not be used upon relapse.49–55 However, if a patient is intolerant to 1 BTK inhibitor, it is appropriate to trial the patient on another BTK inhibitor to potentially improve tolerance.
Follicular lymphoma
Patients with FL commonly present with asymptomatic lymphadenopathy, with waxing and waning symptoms sometimes present for years.56 Asymptomatic patients do not require immediate treatment. However, some will present with more advanced-stage disease or with a greater symptom burden. Indications for treatment include symptomatic disease, threatened end-organ function, cytopenias, massive bulk or splenomegaly at presentation, steady progression over at least 6 months, autoimmune cytopenias, recurrent infections, and/or patient preference.
For patients who require treatment but may not have significant tumor burden or symptoms, rituximab alone or involved-site radiation therapy can be considered as initial therapy, depending on the patient’s age and comorbidities.32 For those who require more intensive upfront treatment, the options include an anti-CD20 monoclonal antibody (mAb) plus chemotherapy or an anti-CD20 mAb plus lenalidomide.32 Both can be followed by anti-CD20 mAb maintenance for 2 years total duration; however, this is not standard practice for all patients.
When patients with relapsed FL require treatment, there are many options, ranging from CD20-mAb alone to combination chemotherapy plus CD20-mAb, radioimmunotherapy, and CD20-mAb plus lenalidomide.32 If FL relapses several years later, options include repeating chemoimmunotherapy, lenalidomide plus rituximab, or radioimmunotherapy, depending on the patient’s age, preferences, and comorbidities. If FL relapses earlier, retreatment with chemoimmunotherapy is not felt to be the best option. Because FL is incurable, many patients will relapse multiple times requiring third, fourth, and beyond lines of treatment. In this situation, the PI3K inhibitors have provided a novel treatment approach for these patients (Table 1).
Idelalisib was the first PI3K inhibitor approved for FL as a single agent.57 This approval was based on a phase 2 trial that included patients with heavily pretreated indolent NHL. Patients with FL (n = 72) who were double refractory after prior exposure to rituximab and an alkylating agent were given idelalisib 150 mg orally twice daily continuously until progression, toxicity, or death. The ORR was 56%, including a 14% CR rate. The median time to response was 2.6 months (range, 1.6–11 months) and the median duration of response was 10.8 months (range, 0–26.9 months).58
Copanlisib was the second PI3K inhibitor available for FL and, unlike idelalisib, is given intravenously weekly rather than orally.59 Copanlisib was studied in the single-arm phase 2 trial CHRONOS-1 that included 104 patients with FL. Similar to idelalisib studies, patients in the copanlisib studies had relapsed after 2 or more lines of therapy. In this study, the ORR was 61% and the CR rate was 16%. The median time to response was 1.7 months (range, 1.3–9.7 months) with a median duration of response of 14.1 months (range, 0–42.5 months).60
Lastly, duvelisib was approved for FL on the basis of a trial (DYNAMO) with similar design to those of copanlisib and idelalisib.61 A total of 83 patients with FL who were both refractory to rituximab and chemotherapy or radioimmunotherapy and who had previously received an alkylating agent or a purine analogue were treated with duvelisib 25 mg orally twice daily. The ORR was 42%, with a CR rate of 1.2%. Here, median time to response was 1.9 months (range, 1.4–11.7 months) with a median duration of response of 10 months (range, 6.3–10.5 months).
Unfortunately, there are no head-to-head comparisons of the available PI3K inhibitors in this space at this time. While cross-trial comparison has inherent limitations, it does not seem that 1 PI3K inhibitor outpaces any other in terms of efficacy. All show similar ORR, time to response, and duration of response; thus, choice of 1 agent over the others should be made after a thoughtful conversation between the patient and medical team, balancing patient preference, financial concerns, and comorbidities in the setting of the various toxicity profiles.
ADVERSE EFFECTS AND MANAGEMENT
Pharmacists are in a unique position to aid in toxicity prevention, monitoring, and management to facilitate improved adherence and optimize outcomes related to treatments for B-cell malignancies. An overview of the toxicity profiles of the novel agents can be found in Table 2, and Table 3 lists drug and comorbidity interactions associated with the novel agents used in lymphoma.
| Table 2: Summary of Adverse Effects of Novel Agents Used in the Treatment of Lymphoma35,36,38,57,59,61-73,75-55,81,84,87-92 |
| Agent |
Class effects |
Comments |
| Ibrutinib |
Atrial fibrillation, bleeding, diarrhea, infection, pneumonia, hypertension, neutropenia, thrombocytopenia, rash, arthralgias/myalgias |
May be associated with greater risk for atrial fibrillation, bleeding, diarrhea, rash, and arthralgias/myalgias than acalabrutinib and zanubrutinib; however, randomized controlled trials comparing the side effect profiles are eagerly awaited |
| Acalabrutinib |
Headache |
| Zanubrutinib |
Neutropenia |
| Idelalisib |
Diarrhea, colitis, infections, hepatotoxicity, pneumonitis, rash |
Black box warnings: hepatotoxicity, diarrhea and colitis, intestinal perforation, infection, pneumonitis |
| Duvelisib |
Black box warnings: diarrhea and colitis, infection, pneumonitis, cutaneous reactions |
| Copanlisib |
No black boxed warnings;
other unique attributes:
hyperglycemia during the infusion,
hypertension during the infusion,
fewer immune-mediated AEs |
| Venetoclax |
TLS, myelosuppression, nausea, diarrhea |
None |
| AE, adverse effects; TLS, tumor lysis syndrome. |
| Table 3: Drug and Comorbidity Interactions with Novel Agents Used in the Treatment of Lymphoma74-76,83,87-89 |
| Agent |
Metabolism |
CYP inhibitors |
CYP inducers |
Renal |
Hepatic |
| Ibrutinib |
CYP3A4 (major),
CYP2D6 (minor) |
Voriconazole: ↓ 140 mg daily?
Posaconazole: ↓ 70 mg daily
Moderate: ↓ 280 mg daily |
Avoid |
No changes |
Child-Pugh A: ↓ 140 mg
Child-Pugh B: ↓ 70 mg
Child-Pugh C: avoid |
| Acalabrutinib* |
CYP3A4 (major),
P-gp, BCRP |
Strong: avoid
Moderate: ↓ 100 mg daily |
↑ 200 mg
twice daily |
No changes |
No changes;
monitor in severe impairment |
| Zanubrutinib |
CYP3A4 (?) |
Strong: ↓ 80 mg daily
Moderate: ↓ 80 mg twice daily |
Avoid |
No changes |
Severe: ↓ 80 mg twice daily |
| Idelalisib** |
CYP3A4 (major),
P-gp, UGT1A4 |
Avoid |
Avoid |
No changes |
Caution
(1.7´ increase AUC) |
| Duvelisib^ |
CYP3A4 (major) |
Strong: ↓ 15 mg daily |
Avoid |
No changes |
No changes |
| Copanlisib |
CYP3A4 (major),
P-gp, BCRP |
Strong: avoid or
↓ 45 mg weekly |
Avoid |
No changes |
Child-Pugh B: ↓ 45 mg
Child-Pugh C: avoid |
| Venetoclax |
CYP3A4 (major),
P-gp |
Strong: avoid during ramp up,
↓ 75% thereafter
Moderate: ↓ 50% |
Avoid |
Monitor TLS |
Monitor toxicity |
*Avoid proton pump inhibitors; take acalabrutinib 2 hours before histamine-2 receptor antagonists and antacids (reduces acalabrutinib AUC 40%–50%).
**Idelalisib inhibits CYP3A4 (strong) and UGT1A1.
^Duvelisib inhibits CYP3A4 (moderate).
Note: all agents listed inhibit P-gp except for acalabrutinib, copanlisib, and duvelisib.
AUC, area under the curve; BCRP, breast cancer resistance protein; CYP, cytochrome P450; P-gp, P-glycoprotein; TLS, tumor lysis syndrome; UGT, UDP-glucuronosyltransferase. |
BTK inhibitors
First-generation BTK inhibitors
Ibrutinib is considered a first-generation BTK inhibitor that, as discussed, greatly changed the treatment paradigm for a wide variety of NHLs. Criticism of ibrutinib, however, stems from its vast and potentially serious toxicity profile.
A pooled analysis of 3 trials utilizing ibrutinib in MCL demonstrated that grade 3/4 adverse effects (AEs) occurred in 80% of patients.35 These grade 3/4 toxicities were most commonly neutropenia (17%), pneumonia (12.7%), thrombocytopenia (12.4%), anemia (10%), AF (6.2%), and hypertension (5.1%).
Unintended binding sites of ibrutinib, as well as its indirect effects on other signaling pathways such as PI3K/AKT, have been proposed as mechanisms of ibrutinib toxicities, particularly AF and hypertension. In general, patients treated with ibrutinib show an increased rate of AF from 4%–10% over what is expected in the general population.62 A pooled analysis of randomized controlled trials for patients with CLL or MCL confirmed the increased risk of AF in ibrutinib-treated patients compared to controls (6% vs. 2% all grades; 3% vs. 1% grade ≥ 3).63 Hypertension has been reported in up to 30% of patients treated with ibrutinib.64 Grade 3/4 hypertension is seen in a smaller percentage of patients (11%); however, ibrutinib-associated hypertension is associated with an increased risk of major adverse cardiovascular events including arrhythmia, MI, stroke, heart failure, and cardiovascular death.65
Safety concerns regarding the combination of ibrutinib and anticoagulant or antiplatelet therapy were raised by investigators during the initial trials: the concerns were based on observations of incidental severe bleeding, including subdural hematomas and post-invasive procedural bleeding.66 Unfortunately, precise information on the number of patients affected and concomitant anticoagulation or antiplatelet therapy was not released. In MCL, grade 3/4 bleeding events occurred in 5.9% of patients total.35 This is similar to what is reported in ibrutinib-based CLL trials, where up to 66% of patients experienced a minor bleeding event and 6% of patients experienced a major bleed.67,68 Of patients experiencing any-grade AF (11.4% of total population), 78.6% of patients received a concomitant anticoagulant and/or antiplatelet medication including warfarin and direct oral anticoagulants (DOACs). Only 1 patient with AF had a major hemorrhage but had not received anticoagulant or antiplatelet medications within 2 years.
This safety profile greatly limited ibrutinib’s utility. Long-term follow-up of early CLL data demonstrated that up to 51% of patients discontinued ibrutinib therapy.69,70 Similarly, a retrospective study of ibrutinib-treated patients in a real-world setting reported a 41% discontinuation rate, with 51% of discontinuations due to ibrutinib-associated toxicities.62
Second-generation BTK inhibitors
Ibrutinib’s off-target effects (through inhibition of tyrosine kinases such as epidermal growth factor receptor, interleukin 2-inducible T-cell kinase, SYK, and TEC) may explain its unique toxicity profile. Because of this, there was interest in developing second-generation BTK inhibitors that demonstrated less off-target inhibition to improve the occurrence of toxicities.
Acalabrutinib was the first second-generation BTK inhibitor to the market, initially approved for MCL and subsequently approved for CLL. Acalabrutinib is more highly-selective to the BTK receptor and shows very little to no inhibition of various off-target enzymes. Clinical trial data with acalabrutinib demonstrated significant improvement in the toxicity profile, with the most commonly reported any-grade AEs being headache (39%), diarrhea (31%), fatigue (28%), myalgia (21%), and bruising (21%). Headache events were mostly grade 1 (64%), and the majority of patients (77%) reported only 1 event. Headaches occurred early in treatment, at a median onset of 5 days, and were manageable.36 Only 4% of patients withheld therapy due to headache and no patients permanently discontinued acalabrutinib due to this AE. The most frequently reported grade 3/4 AEs were neutropenia (15%) and anemia (10%). In this early MCL analysis, there were no cases of AF. Subsequent trials of acalabrutinib in CLL demonstrated that AF occurs in up to 5% of patients.30,71 Three (2%) grade 3/4 cardiac AEs were reported in 1 patient each; however, these events were found to be unrelated to acalabrutinib therapy.36 Bleeding events, the most frequent of which were contusion and petechiae, occurred in 39 (31%) patients and were all grade 1 or 2, except for 1 grade 3 gastrointestinal bleed. While the rate of grade 3 hemorrhage is low for acalabrutinib, the true incidence is around 3% of treated patients.71 One patient developed grade 3 hypertension during acalabrutinib therapy.
Zanubrutinib also has greater specificity to the BTK receptor and less off-target inhibition than ibrutinib. There are, however, minor differences in the off-target profiles between zanubrutinib and acalabrutinib, which likely explain small variances in their toxicity profiles. In the pivotal trial that gained zanubrutinib approval in MCL, the most common any-grade AEs were diarrhea (35%), petechiae (31%), upper respiratory tract infection (27%), fatigue (25%), constipation (21%), rash (19%), headache (17%), and peripheral edema (17%).38 Grade 3/4 AEs occurred in 58.3% of patients overall and were most frequently anemia (11%), thrombocytopenia (9%), neutropenia (9%), and pneumonia (6%). Severe hemorrhage, grade 3 hypertension, and AF occurred in 3% of patients each. In all, 23% of patients discontinued zanubrutinib due to an AE.
While comparative cross-trial analysis of the 3 available BTK inhibitors demonstrates remarkable improvements in the toxicity profile for second-generation BTK inhibitors, a head-to-head comparison demonstrating this improvement was lacking. Recently, results of the randomized phase 3 ASPEN trial that compared zanubrutinib to ibrutinib in WM patients were presented.47 Patients treated with zanubrutinib showed lower rates of AF (2% vs. 15%), major bleeding (6% vs. 9%), diarrhea (21% vs. 32%), peripheral edema (9% vs. 19%), muscle spasms (10% vs. 24%), hypertension (11% vs. 17%), and pneumonia (2% vs. 12%) despite including an older patient population (age > 75 years, 33% zanubrutinib vs. 22% ibrutinib). Additionally, AEs leading to discontinuation or death were lower with zanubrutinib than with ibrutinib. Zanubrutinib, however, did show higher rates of neutropenia (25% vs. 12%) and anemia (12% vs. 10%). The increased incidence of neutropenia did not translate into an increased risk of grade 3/4 infection (18% vs. 19%).
On the basis of these results, clinicians are becoming increasingly more comfortable with second-generation BTK inhibitors due to similar efficacy and improved toxicity profiles. It is important to note, however, that while the second-generation BTK inhibitors show lower incidence of major ibrutinib toxicities, they are not completely without risks. Thus, the guiding principles for management and monitoring of these major toxicities applies to all 3 available agents.
Patient case 2: PA is a 68-year-old female who has R/R MCL that is being treated with ibrutinib 560 mg orally daily. She presents to her 3-month follow-up clinic visit and, upon interrogation by the clinic pharmacist, she admits to periodically feeling as though “her heart is beating outside of her chest.” She is not bothered by it, but does find it to be annoying as it comes and goes.
Management of BTK inhibitor toxicities
Atrial fibrillation
In general, the risk of developing BTK inhibitor-associated AF is highest during the first 3 months of therapy, with a median time to onset of 2.8 months; however, late-onset AF also occurs.63 Identified risk factors for ibrutinib-related AF include older age (≥ 65 years), male sex, hypertension, and a history of pre-existing cardiac disease and AF, diabetes mellitus, and valvular heart disease.72
It should be noted that previous history of AF or new development of AF on a BTK inhibitor is not a contraindication to BTK inhibitor therapy continuation. BTK inhibitors can be safely given to patients with AF.73 Of patients with a history of AF or an arrhythmia, 70% will have no recurrence on ibrutinib therapy.35
Management of AF in a patient receiving a BTK inhibitor requires a collaborative approach between the treating hematologist and cardiologist and should balance the patient’s individual risk of stroke versus the competing risk of bleed (including BTK inhibitor-associated bleeding). For rate and rhythm control, diltiazem, verapamil, and amiodarone are inhibitors of cytochrome P450 (CYP) 3A4, which is a primary site for ibrutinib, acalabrutinib, and zanubrutinib metabolism. Concurrent use may increase the serum BTK inhibitor concentration and potentially increase toxicity.74–76 Additionally, serum amiodarone concentration may be increased secondary to ibrutinib’s inhibition of P-glycoprotein (P-gp), leading to increased toxicity. Non-interacting medications for rate and/or rhythm control should be utilized when able. If an interacting medication cannot be avoided, appropriate BTK inhibitor dose adjustments should be considered, balancing patient disease control and potential risk of toxicities.
In patients at high risk for systemic thromboembolic events (specifically stroke) according to CHADSVASC score, the most appropriate choice of an anticoagulant for BTK inhibitor-associated AF is unknown. Consideration must be given to interactions between P-gp inhibitors (ibrutinib) and substrates (dabigatran, digoxin) and CYP3A4 inhibitor antiarrhythmic drugs and DOACs (rivaroxaban, apixaban). In general, DOACs are preferred over vitamin K antagonists due to competing bleed risks.74–76
In patients with uncontrollable AF on BTK inhibitor therapy, optimal management is unknown: consideration should be given to discontinuation of the BTK inhibitor. For patients on ibrutinib with uncontrollable or recurrent AF, switching to a second-generation BTK inhibitor can be considered due to the lower reported incidence of AF. However, further data are needed to understand if switching from a first- to second-generation BTK inhibitor after development of BTK inhibitor-associated AF truly reduces the risk of AF recurrence.72
Patient case 2 (continued): An electrocardiogram is performed on PA, which reveals AF with a rapid heart rate. She is considered to have grade 2 AF. After discussion with her primary hematologist, you decide to start PA on metoprolol 25 mg orally twice daily and aspirin 81 mg orally daily and to continue the ibrutinib without a dose reduction. You counsel PA on her new medications and provide her with a management plan for when to call the clinic versus when to present to the nearest emergency room.
PA’s hematologist places electronic prescriptions to be sent to her local pharmacy. While placing these orders, an alert appears warning the hematologist about an interaction between ibrutinib and aspirin. They look to you for guidance.
Bleeding and anticoagulation
Bleeding in patients on BTK inhibitors is thought to be due to both on-target and off-target kinase inhibition. BTK, along with additional members of the SFK (src family kinases) and TEC families, have a known role in several platelet activation and adhesion functions.77 When BTK is inhibited, TEC can compensate, resulting in almost fully functioning platelet activation and adhesion. When concurrent TEC inhibition occurs, platelet aggregation and thrombus formation and stability are greatly inhibited.
Ibrutinib shows strong inhibition of multiple players involved in the thrombus-formation cascade, including inhibition of BTK and TEC. Unlike ibrutinib, second-generation BTK inhibitors (acalabrutinib and zanubrutinib) do not inhibit TEC. Therefore, the impact of BTK inhibition alone leads to only mildly diminished platelet activation and results in much more stable thrombus formation. The lower rate of major and minor bleeding events for the second-generation BTK inhibitors are likely due to these differences in on- and off-target effects. Because of this difference in risk, in patients for whom the risk of a major or minor bleed is felt to be elevated at baseline, second-generation BTK inhibitors offer an attractive treatment approach.
It is recommended to avoid the combination of warfarin and BTK inhibitor therapy due to the heightened risk of fatal bleeds noted in early clinical trials.66 However, use of anticoagulation or antiplatelet therapy in patients on BTK inhibitors is not a contraindication. Clinical trial data regarding concurrent use of anticoagulant and/or antiplatelet therapy in patients on BTK inhibitors are limited. Data with acalabrutinib in MCL showed concurrent anticoagulant use was reported in 46% of patients and no patients on concurrent anticoagulant therapy experienced a grade ≥ 3 bleeding event.37 Of 39 patients who had bleeding events, 4 had concurrent use of either aspirin or clopidogrel at the time of the event. These 4 patients had grade 1 or 2 petechiae, contusion, or purpura. The incidence of major bleeding (grade ≥ 3) in real-world patients on ibrutinib therapy is 18%, with 54% of these major bleeds occurring in patients on both an antiplatelet and anticoagulant medication.78 Patients on antiplatelet therapy alone accounted for 30% of bleeds, whereas those on an anticoagulant alone showed an 8% risk of major bleed.
When considering the combination of BTK inhibitor and antiplatelet therapy, it is important to note that dual antiplatelet therapy (DAPT) can increase the risk of major bleeding by 40%–50% compared to single antiplatelet therapy.79 Discussion surrounding the patient’s clinical scenario and risk of bleed based on generation of BTK inhibitor should occur in the presence of DAPT, and single antiplatelet therapy should be utilized when possible. DOACs are likely a safer class of anticoagulants to combine with BTK inhibitors than warfarin. As always, the risks and benefits of anticoagulation should be considered. When considering the combination of DOACs and BTK inhibitors, it is important to note BTK inhibitor and DOAC (apixaban and rivaroxaban) metabolism via CYP3A4. While it is unknown how the presence of 2 CYP3A4 substrates impacts metabolism, clinicians should remain prudent of interacting CYP3A4 inhibitor medications, such as those used for AF, as increased exposure to both BTK inhibitor and DOAC can be seen.74–76 As a general rule, caution, close follow-up, and patient education should be utilized in patients who require anticoagulation or antiplatelet therapy while on BTK inhibitor therapy.
Patient case 2 (continued): You respond: the combination of BTK inhibitors and antiplatelet therapies increases the risk of major and minor bleeds. Of the BTK inhibitors, ibrutinib may have the highest bleed risk according to ibrutinib’s off-target inhibition of TEC and SRC; however, randomized controlled trials are needed to confirm this observation. Continuation of a BTK inhibitor in the setting of antiplatelet agents and/or anticoagulation (except for warfarin) is not a contraindication.
PA now presents to clinic for her 1-year follow-up. She states she is doing quite well except for continued hypertension despite her primary care physician maximizing her metoprolol dose. The hematologist asks you whether they should stop ibrutinib and switch to a different BTK inhibitor or switch to a venetoclax-based regimen because of PA’s uncontrollable hypertension.
Hypertension
New onset or worsening of pre-existing hypertension is another notable adverse event of BTK inhibitors. Unlike most other BTK inhibitor toxicities, the incidence of hypertension remains stable over time.67,68 If hypertension occurs, BTK inhibitor therapy can be continued and the hypertension is medically managed in conjunction with the patient’s primary care or cardiology provider.
Adequate management of BTK inhibitor-associated hypertension with antihypertensives seems to mitigate the risk of a major cardiac event.65 Patients with uncontrolled hypertension despite multiple antihypertensives should be considered for a dose reduction and/or BTK inhibitor discontinuation to prevent long-term cardiovascular sequelae.65 In patients on ibrutinib who demonstrate uncontrollable hypertension, consideration can be given to switching to a second-generation BTK inhibitor, which are associated with a lower incidence of hypertension.
Patient case 2 (continued): BTK inhibitor-associated hypertension increases the risk for a major cardiovascular event. These cardiovascular events may be mitigated by controlling PA’s blood pressure. While she is on 1 maximized antihypertensive, adding an additional agent for blood pressure control would be appropriate at this time. Ibrutinib therapy should only be stopped if PA’s hypertension remains uncontrolled despite multiple maximized antihypertensives. Acalabrutinib is also associated with hypertension and it is difficult to say at this time whether switching to a second-generation BTK inhibitor would abrogate the hypertension. Further guidance from randomized trials comparing the 2 agents could reveal the optimal strategy. Abandoning the BTK inhibitor class that is controlling PA’s MCL for a BCL2 inhibitor would be inappropriate until PA’s hypertension was deemed uncontrollable despite reasonable attempts.
Additional toxicities
Arthralgia and myalgia. The exact mechanism of arthralgias and myalgias secondary to BTK inhibitor therapy remains unknown. As with other toxicities, the risk seems to be more pronounced with ibrutinib therapy and decreased with second-generation agents. In general, arthralgia may be more pronounced early in the treatment course and resolve over time.67 However, there are reports of late-onset arthralgia. Management techniques utilize acetaminophen or short pulses of corticosteroids, if severe.76 It is recommended to avoid anti-inflammatory agents with antiplatelet properties due to the secondary risk of bleeding. For patients whose quality of life is severely diminished by arthralgias, BTK inhibitor therapy can be withheld and restarted at a lower dose. Additionally, transition to an alternative BTK inhibitor, if the patient is on ibrutinib, may result in arthralgia resolution.80
Diarrhea. Diarrhea is a common AE that will impact almost 50% of patients treated with ibrutinib and 20%–30% of patients treated with acalabrutinib or zanubrutinib. BTK inhibitor-associated diarrhea is rarely severe. Its incidence is highest within the first 6 months of therapy and declines rapidly over time, with a median duration of 6–20 days.67,68,81 In general, because the diarrhea is self-limiting, it will resolve without intervention or with minimal dietary intervention. In cases of moderate-severe diarrhea, infectious etiology should be ruled out. Otherwise, standard antidiarrheals such as loperamide should be utilized. In patients with more severe diarrhea, BTK inhibitor therapy can be withheld until diarrhea resolves and resumed at a reduced dose.76
Rash. Rash with BTK inhibitors is usually self-limiting and does not require dose reduction or holds. Management should be determined according to the severity of the rash (i.e., the extent of body surface area impacted) and patient symptoms. In general, patients can be treated with topical steroids and antihistamines while therapy is continued.76 For patients who fail to respond or who present with more severe rash, the BTK inhibitor can be held and oral corticosteroids may be utilized. BTK inhibitor therapy discontinuation due to rash is extremely rare.
Headache. Headache is a unique toxicity that patients may experience while initiating acalabrutinib.75 For most patients, headache abates over time with no pharmacological interventions. If treatment is required, acetaminophen, caffeine, and/or hydration are modalities that may be effective. Patients are counseled to avoid ibuprofen and other non-steroidal anti-inflammatory agents due to the risk of bleeding, unless a discussion of the risks versus benefits has occurred with their treating hematology team.
Management of BCL2 inhibitor (venetoclax) toxicities
Tumor lysis syndrome
TLS may be referred to as the most critical toxicity of venetoclax, contributing to treatment-related death in early CLL clinical trials. The currently utilized ramp-up dosing schedule was implemented to mitigate the TLS risk noted in these early analyses.82 Optimal avoidance and management of TLS related to venetoclax therapy should be determined on the basis of an assessment of the patient’s risk for TLS according to disease burden as well as the patient’s clinical factors such as age and kidney function.83
Patients with NHL receiving venetoclax can be stratified into low-, intermediate-, and high-risk categories for treatment-related TLS primarily on the basis of assessment of tumor burden (measured by lymph node size and blood counts) and reduced renal function (measured by creatinine clearance) (Table 4).84 While this assessment tool was developed by examining severe or fatal cases of TLS from early CLL clinical trials, it is utilized broadly for other NHLs, including MCL, as high lymphocyte counts have been associated with fatal TLS in these patients, as well.85
| Table 4: Venetoclax TLS Management84 |
TLS risk |
Disease characteristics* |
Management plan |
Low |
No bulky adenopathy
ALC < 25 ´ 109/L |
Outpatient:
· Oral hydration (1.5–2 L per day) and allopurinol
· Lab monitoring: pre-dose and 6–8 hours & 24 hours after first dose of 20 mg and 50 mg and then pre-dose at subsequent ramp-up doses |
Intermediate |
Bulky adenopathy:
≥ 5 cm and < 10 cm
or
ALC ≥ 25 ´ 109/L |
Outpatient:
· Oral hydration (1.5–2 L per day), IV hydration (PRN), and allopurinol
· Lab monitoring: pre-dose and 6–8 hours & 24 hours after first dose of 20 mg and 50 mg and then pre-dose at subsequent ramp-up doses
· If creatinine clearance < 80 mL/min, consider inpatient admission for first 2 dose escalations |
High |
Bulky adenopathy: ≥ 10 cm
or
Bulky adenopathy: ≥ 5 cm and ALC ≥ 25 ´ 109/L |
· Inpatient for first dose of first 2 ramp-up doses
· Oral hydration (IV as tolerated)
· Allopurinol (consider rasburicase on the basis of baseline uric acid) |
*Additional factors to consider in risk assessment: baseline uric acid, LDH, potassium, phosphorous, serum creatinine, and calcium.
ALC, absolute lymphocyte count; IV, intravenously; LDH, lactate dehydrogenase; PRN, as needed; TLS, tumor lysis syndrome. |
To reduce the risk of laboratory and clinical TLS, there are several steps and interventions that should be utilized for prevention and management. After initial patient evaluation of TLS risk, monitoring can occur on either an inpatient (high risk) or outpatient (low to intermediate risk) basis. Management and prophylaxis utilizes intensive oral or intravenous hydration and administration of antihyperuricemic agents to lower uric acid levels. Consideration can also be given to the use of potassium- and phosphate-reducing agents, as necessary.
Data demonstrate that when venetoclax dose escalation and TLS risk management protocols are utilized, clinical manifestations of venetoclax-associated TLS can be mitigated.86 It is important to note that the 5-week dose escalation strategy of venetoclax is designed to gradually debulk tumor burden and decrease risk of TLS. Because of this, management and mitigation protocols are generally followed for the first 2 venetoclax dose escalations (20 mg and 50 mg), with the risk of TLS significantly declining for subsequent escalations. For patients who require inpatient monitoring according to risk, generally only the first 2 dose escalations need to occur as an inpatient. However, the TLS monitoring and mitigation plan following the first 2 dose escalations should be determined on a case-by-case basis according to patient clinical scenario and risk factors.
In patients with laboratory signs of TLS, venetoclax should be held.83 The dose can be continued if the TLS resolves within 24–48 hours. The venetoclax dose should be reduced if the TLS persists for more than 48 hours, once laboratory values stabilize. Once venetoclax titration is complete, the ongoing risk of TLS is minimal and clinically evident TLS is rare. Prophylactic measures such as aggressive oral hydration, antihyperuricemics, phosphate binders, and potassium-reducing agents can be discontinued at that time.
Neutropenia
While TLS is the most feared AE, neutropenia is the most common grade 3/4 toxicity reported with venetoclax therapy. Grade 3/4 neutropenia with venetoclax may impact up to 61% of patients.84 Patients should be monitored with frequent blood count assessments and for signs of infection, and patient education about this increased risk should be provided. While the risk is high, rates of febrile neutropenia are low, occurring in ≤ 5% of patients.86
Patients with neutropenia may require omission of a venetoclax dose or a dose reduction but can often be managed with growth factor support. It is currently recommended to withhold venetoclax for grade 3 neutropenia with signs of infection or fever and for grade 4 neutropenia. Venetoclax can be resumed at the same dose when the AE resolves to grade 1 or with resolution of infection.83 Patients can receive growth factor support at the provider’s discretion. Management of subsequent occurrences includes treatment interruption, ongoing use of growth factor support, and resumption of venetoclax at a lower dose when neutropenia resolves.
Management of PI3K inhibitor toxicities
The 3 available PI3K inhibitors (idelalisib, duvelisib, and copanlisib) differ according to their inhibition of the various PI3K isoforms. These differences in isoform inhibition also contribute to various differences in their toxicities profiles.
Idelalisib targets the δ form of PI3K. Common, any-grade AEs of idelalisib include neutropenia (56%), diarrhea (43%), elevated transaminases (35%–47%), nausea (30%), fatigue (30%), cough (29%), and pyrexia (28%).57 Common severe toxicities include diarrhea (13%), pneumonia (7%), neutropenia (27%), and increased alanine transaminase (ALT) (13%). Diarrhea can represent an immune-mediated colitis in 3% of patients. Febrile neutropenia occurs in 3% of patients treated with idelalisib and 21% of patients will experience a fatal or serious infection. Pneumonitis is seen in about 2% of patients.87
Similar to idelalisib, duvelisib also targets PI3K-δ but inhibits the γ isoform as well. For the most part, the toxicity profiles of duvelisib and idelalisib overlap. The most common any-grade toxicities reported with duvelisib include diarrhea (48.8%), nausea (29.5%), neutropenia (28.7%), fatigue (27.9%), and cough (27.1%).61 The most common grade 3/4 toxicities are neutropenia (24.8%), diarrhea (14.7%), anemia (14.7%), thrombocytopenia (11.6%), and pneumonia (5.4%). Immune-mediated colitis occurs in up to 8% of patients and pneumonitis in 5%. Hepatic injury, as evident by transaminase elevation occurs less frequently (ALT increase: any-grade, 14%; grade 3/4, 5.4%). Febrile neutropenia occurs in 9.3% of patients and 31% of those treated with duvelisib will experience a fatal or serious infection. Inhibition of PI3K-γ may explain the higher rate of myelosuppression and infection seen with duvelisib due to inhibition of an additional PI3K isoform involved in immune regulation.88
Copanlisib, unlike duvelisib and idelalisib, inhibits PI3K-α in addition to the δ and γ isoforms. In general, toxicities commonly observed with idelalisib and duvelisib are less common and less severe with copanlisib. Diarrhea occurs in 34% of patients; however, only 5% of patients will have severe diarrhea and 1% of patients will experience immune-mediated colitis.59 Common any-grade toxicities with copanlisib include fatigue (30%), fever (25%), nausea (23%), pneumonia (21%), cough (16%), elevated transaminases (23%–28%), and myelosuppression (20%–30%). Only 1% of patients will experience severe hepatic injury and 8% will experience pneumonitis. Due to its inhibition of PI3K-δ , copanlisib retains an infection risk and 19% of patients will experience a fatal or serious infection.89 Two of the most common toxicities of copanlisib unique to this agent and not seen with other PI3K inhibitors include hyperglycemia and hypertension. These are likely related to copanlisib’s inhibition of PI3K-α. Hyperglycemia occurs in up to 50% of patients and is severe in 30%–40% of patients. Any-grade hypertension is reported in up to 30% of patients and is generally more severe in nature (24%).
Because of their toxicity profiles, both duvelisib and idelalisib carry black box warnings for diarrhea, colitis, infection, and pneumonitis.87,88 Additional black box warnings include hepatotoxicity and intestinal perforation for idelalisib and cutaneous reactions for duvelisib. Because of copanlisib’s less severe toxicity profile, it does not have any black box warnings at this time; however, hypertension and hyperglycemia appear to be its toxicities of greatest concern. The PI3K inhibitor recommended should be highly dependent on the AE profile and patient clinical scenario after discussion of the risks and benefits with the patient. Likely the choice of a specific PI3K inhibitor will depend on patient wishes, comorbidities, performance status, and convenience. Patients should be counseled on the vast but potentially serious AE profiles of the different PI3K inhibitors with a close monitoring and management plan in place prior to therapy initiation.
Diarrhea and colitis
Diarrhea can be categorized into 2 groups: the first is typically self-limiting, responds well to anti-motility agents, and occurs within the first 8 weeks of initiation of therapy (median time of onset is 1.5 months)90; the second type occurs at a median time of 7.1 months after beginning treatment, responds poorly to anti-motility agents, and tends to be more severe in nature.91
Consensus guidelines have been created for both work-up and management of PI3K inhibitor-associated early and late diarrhea.90 While these guidelines are specific to idelalisib, they were created prior to approval of duvelisib and can be utilized across the various PI3K inhibitors. Management includes infectious work up, dietary modifications, and anti-motility agents, if appropriate. For mild diarrhea, therapy can generally be continued with close patient monitoring and follow-up. For severe diarrhea, PI3K inhibitor therapy should be withheld until resolution and restarted at a lower dose. Corticosteroids can be utilized for persistent diarrhea or immune-mediated colitis after infectious causes are ruled out. For late-onset diarrhea, management follows the same guiding principles. For patients with recurrent grade 3 diarrhea or for those who experience life-threatening diarrhea or colitis, PI3K inhibitor therapy should be permanently discontinued.
Infection
The PI3K pathway plays an important role in regulating the immune system and a large percentage of patients will experience a fatal or life-threatening infection with PI3K inhibitors, irrespective of agent. Among infectious complications noted in clinical trials, a small percentage of patients developed Pneumocystis jirovecii pneumonia (PJP) and cytomegalovirus (CMV) infections (roughly 1% of patients each).57,59,61,87–89 Because of this, all patients should receive appropriate PJP prophylaxis during the entire duration of PI3K inhibitor therapy. Clinical and laboratory monitoring for CMV infection is recommended for those with a history of CMV infection or who demonstrate positive CMV serology prior to the start of treatment. For any suspected or confirmed infection, PI3K inhibitor therapy should be withheld until infectious symptoms or infection has resolved but can be restarted upon resolution. However, given the high rate of infectious complications with PI3K inhibitors, clinicians should balance clinical efficacy with ongoing infection risk in patients who experience an initial infectious complication. Consideration should be given to permanent PI3K inhibitor therapy discontinuation for these patients. In the case of confirmed PJP infection, PI3K inhibitor therapy should be permanently discontinued.
Pneumonitis
While pneumonitis is a rare complication of PI3K inhibitor therapy, it is a serious complication that can have long-term negative consequences on patient outcomes. Comprehensive patient education on concerning symptoms should be provided in order to prompt appropriate medical management: patients should be instructed to monitor for symptoms including cough, dyspnea, hypoxia, and fever and to seek quick medical care if they occur. Patients with suspected PI3K inhibitor-associated pneumonitis should discontinue the drug and undergo complete infectious work-up.87–89 Chest imaging may demonstrate interstitial infiltrates. If infectious etiology is negative and patient fails to improve on antimicrobial therapy, treatment with corticosteroids should be considered. For patients with suspected pneumonitis, rechallenge with PI3K inhibitor should be avoided.
Transaminitis
Aspartate transaminase (AST) and ALT elevations occur most commonly in the first 12 weeks of therapy and are generally reversible with therapy interruption. However, up to 26% of patients will experience recurrent transaminitis even after dose reduction upon rechallenge.90 Liver function tests (LFTs) should be monitored as often as every 2 weeks during the first 3 months of therapy when the risk is highest, followed by monthly monitoring as therapy continues. Patients can continue on therapy with AST/ALT elevations up to 3 to 5 times the upper limit of normal (ULN). For patients who experience laboratory transaminitis, LFTs should be monitored weekly until resolved. For patients who experience more severe elevations (5–20´ ULN), therapy should be withheld and resumed at a lower dose once normalized. For those with AST/ALT > 20 times ULN or who experience life-threatening or permanent liver damage, therapy should be permanently discontinued.
Hypertension
Hypertension associated with copanlisib usually occurs early (after cycle 1) and peaks at 2 hours post-infusion.89,92 Blood pressure (BP) will begin to decrease at 2 hours post-infusion and full resolution is typical within 24 hours.93 Increases in systolic and diastolic BP are on average 16.8 mmHg and 7.8 mmHg, respectively.89 It should be noted that serious hypertensive events requiring hospitalization are rare (0.9%) and no patients in clinical trials experienced grade 4 hypertension (i.e., life-threatening complication, hypertensive urgency).
At minimum, BP should be monitored pre- and post-infusion.89 Patients with pre-existing hypertension should have this managed and controlled with an optimized antihypertensive management plan prior to copanlisib therapy initiation, if possible.94 Copanlisib should be withheld until pre-dose BP is < 150/90 mmHg. For those with pre-dose BP ≥ 150/90 mmHg, therapy should be withheld until BP is within goal on at least 2 consecutive occasions at least 15 minutes apart.
For patients with post-dose BPs ≥ 150/90 mmHg with non-life-threatening symptoms, the decision of whether to treat with antihypertensives is based on patient-specific risk factors (e.g., baseline BP, severity of elevation, pre-existing cardiovascular risk factors such as diabetes or chronic kidney disease, or cardiovascular disease such as stroke, heart failure, or peripheral vascular disease). Patients who demonstrate life-threatening symptoms should be managed appropriately and copanlisib should be permanently discontinued. For patients who demonstrate post-copanlisib BP < 150/90 mmHg without use of antihypertensive therapies, no dose adjustment of copanlisib needs to be made for future infusions.
If BP treatment during or after the copanlisib infusion is planned, short-acting antihypertensives should be used given the short anticipated duration of hypertension from copanlisib treatment. For any patient in whom BP remains uncontrolled despite appropriate at-home antihypertensive therapies, the dose of copanlisib can be reduced. If the BP remains elevated after appropriate dose reductions, copanlisib therapy should be discontinued.
Hyperglycemia
Hyperglycemia with copanlisib is common but tends to be transient, occurring during the infusion, and is manageable. Maximum changes from baseline in plasma glucose occur at 5 hours after the infusion, returning to baseline within 24 hours. Patients tend to remain asymptomatic from these blood glucose (BG) elevations.92 Permanent BG elevations from baseline will occur in approximately 18% of patients, and only 10% of patients treated with copanlisib will develop hemoglobin A1c (HbA1c) values of > 6.5%.59 Hyperglycemia leading to dose reductions and drug discontinuations occurs in 7% and 2% of patients, respectively.89
For most patients, acute hyperglycemia due to PI3K inhibition should not lead to immediate discontinuation. Dose modifications or discontinuation for hyperglycemia should only be considered in situations of severe events or if hyperglycemia persists after therapeutic intervention.
BG should be optimized in all patients prior to initiation of copanlisib therapy, if possible, especially in those with a history of diabetes or prediabetes. Based on experience from clinical trials, current recommendations are to delay therapy in patients with a pre-infusion fasting BG of ≥ 160 mg/dL or a random blood glucose of ≥ 200 mg/dL. For patients with a post-dose BG ≥ 500 mg/dL, on first occurrence, copanlisib should be withheld until the fasting glucose is ≤ 160 mg/dL or the random BG is ≤ 200 mg/dL. In patients who meet appropriate pre-infusion BG parameters, BG should be monitored during the infusion, according to an institution-based protocol and patient risk. BG monitoring during the copanlisib infusion and post-infusion may not need to occur for non-diabetic patients.
During initial phase 1 trials, diabetic patients receiving copanlisib were managed with short-acting insulin and/or oral glucose-lowering agents following the infusion.93 In the phase 2 studies for indolent NHL, including FL, rapid, short-acting insulin was recommended per the investigator’s discretion.95 If a patient experienced recurrent hyperglycemia or prolonged elevated BG with subsequent infusions, investigators could recommend the use of intermediate or long-acting insulin or an oral hypoglycemic agent. In the phase 2 CHRONOS-1 study, hyperglycemia during treatment was managed with rapid- or short-acting insulin with a 3-hour observation period after the administration of insulin to monitor for hypoglycemia.59 Insulin-naïve patients could also receive intravenous hydration as an insulin-sparing approach. While no hypoglycemic events have been reported, concern remains regarding the risk of hypoglycemia with the use of insulin for glycemic control, as the duration of hyperglycemia may be shorter than the time required for insulin to act.
As a general approach, diabetic or prediabetic patients should consider consulting with an endocrinologist or their primary care provider prior to the start of copanlisib treatment if time permits. Patients may continue on their current antidiabetic medication regimens with close monitoring for adjustments. If medications need to be added or changed, consideration should be given to oral hypoglycemics that do not cause hypoglycemia. For patients who are insulin dependent and for whom oral alternatives are not an option, frequent insulin adjustments should not be made: adjustments should be made after a trend is noted with BGs following copanlisib infusions. It seems that, similar to non-diabetic patients, only a small percentage of patients with diabetes will have a long-lasting impact on their overall diabetes control. In diabetic patients treated with short-acting insulin and/or oral glucose-lowering agents following copanlisib infusions, the mean baseline HbA1c was 7.4%, which changed to 7.3% after treatment with copanlisib.93
CONCLUSION
The advent of several novel targeted therapies has changed the treatment landscape for B-cell malignancies. These advances have dramatically improved patient outcomes and tolerability to therapy. Despite the success of BTK inhibitors, PI3K inhibitors, and the BCL2 antagonist, there are still barriers that prevent the optimal use of these therapies for patients and clinicians. Many patients will discontinue these novel therapies due to toxicities or financial barriers. Pharmacists play integral roles working as part of the multidisciplinary team, including learning from, teaching, and helping navigate patients and the team through the safe and effective integration of these novel agents into patient care. Pharmacists are also in a unique position to aid in toxicity prevention, monitoring, and management to facilitate improved adherence and optimize outcomes.
REFERENCES
- Al-Hamadani M, Habermann TM, Cerhan JR, et al. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: a longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am J Hematol. 2015;90(9):790–5.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
- Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013;34(12):592–601.
- Kraus M, Alimzhanov MB, Rajewsky N, Rajewsky K. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117(6):787–800.
- Scupoli MT, Pizzolo G. Signaling pathways activated by the B-cell receptor in chronic lymphocytic leukemia. Expert Rev Hematol. 2012;5(3):341–8.
- Stevenson FK, Krysov S, Davies AJ, et al. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313–20.
- Kaptein A, de Bruin G, Emmelot-van Hoek M, et al. Potency and selectivity of BTK inhibitors in clinical development for B-cell malignancies. Blood. 2018;132(Supplement 1):1871.
- Sopasakis VR, Liu P, Suzuki R, et al. Specific roles of the p110α isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010;11(3):220–30.
- Jackson SP, Schoenwaelder SM, Goncalves I, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11(5):507–14.
- Sasaki T, Irie-Sasaki J, Jones RG, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287(5455):1040–6.
- Okkenhaug K, Bilancio A, Farjot G, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031–4.
- Lannutti BJ, Meadows SA, Herman SEM, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–4.
- Marini BL, Samanas L, Perissinotti AJ. Expanding the armamentarium for chronic lymphocytic leukemia: a review of novel agents in the management of chronic lymphocytic leukemia. J Oncol Pharm Pract. 2017;23(7):502–17.
- Göckeritz E, Kerwien S, Baumann M, et al. Efficacy of phosphatidylinositol-3 kinase inhibitors with diverse isoform selectivity profiles for inhibiting the survival of chronic lymphocytic leukemia cells. Int J Cancer. 2015;137(9):2234–42.
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63.
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–83.
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–64.
- Llambi F, Moldoveanu T, Tait SWG, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44(4):517–31.
- Anderson MA, Huang D, Roberts A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin Hematol. 2014;51(3):219–27.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. v4.2020. https://www.nccn.org/professionals/physician_gls/pdf/cll.pdf. Published December 20, 2019. Accessed July 18, 2020.
- National Cancer Institute. Cancer Stat Facts: Leukemia – Chronic Lymphocytic Leukemia (CLL). https://seer.cancer.gov/statfacts/html/clyl.html. Accessed July 18, 2020.
- Cramer P, Hallek M. Prognostic factors in chronic lymphocytic leukemia-what do we need to know? Nat Rev Clin Oncol. 2011;8(1):38–47.
- Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2015;127(2):208–15.
- Tam CS, O’Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112(4):975–80.
- Thompson PA, Tam CS, O’Brien SM, et al. Fludarabine, cyclophosphamide, and rituximab achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood. 2016;127(3):303–9.
- Rossi D, Terzi-di-Bergamo L, De Paoli L, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015;126(16):1921–4.
- Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–43.
- Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517–28.
- Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225–36.
- Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278–91.
- Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol. 2019;94(6):710–25.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: B-Cell Lymphomas. v3.2020. https://www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf. Accessed August 20, 2020.
- Le Gouill S, Thieblemont C, Oberic L, et al; LYSA Group. Rituximab after autologous stem-cell transplantation in mantle-cell lymphoma. N Engl J Med. 2017;377(13):1250–60.
- Gordon LI, Schieber M, Karmali R. Current overview and treatment of mantle cell lymphoma. F1000Res. 2018;7:F1000 Faculty Rev-1136.
- Rule S, Dreyling M, Goy A, et al. Ibrutinib for the treatment of relapsed/refractory mantle cell lymphoma: extended 3.5-year follow up from a pooled analysis. Haematologica. 2019;104(5):e211–4.
- Wang M, Rule S, Zinzani PL, et al. Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2018;391(10121):659–67.
- Wang M, Rule S, Zinzani PL, et al. Durable response with single-agent acalabrutinib in patients with relapsed or refractory mantle cell lymphoma. Leukemia. 2019;33(11):2762–6.
- Song Y, Zhou K-S, Zou D, et al. Treatment of patients with relapsed or refractory mantle-cell lymphoma with zanubrutinib, a selective inhibitor of Bruton’s tyrosine kinase. Clin Cancer Res. 2020. Online ahead of print.
- Davids MS, Roberts AW, Seymour JF, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol. 2017;35(8):826–33.
- Eyre TA, Walter HS, Iyengar S, et al. Efficacy of venetoclax monotherapy in patients with relapsed, refractory mantle cell lymphoma after Bruton tyrosine kinase inhibitor therapy. Haematologica. 2019;104(2):e68–71.
- Tam CS, Anderson MA, Pott C, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378(13):1211–23.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Waldenström Macroglobulinemia/Lymphoplasmacytic Lymphoma. v2.2020. https://www.nccn.org/professionals/physician_gls/pdf/waldenstroms.pdf. Published April 15, 2020. Accessed July 19, 2020.
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med. 2012;367(9):826–33.
- Baron M, Simon L, Poulain S, Leblond V. How recent advances in biology of Waldenström’s macroglobulinemia may affect therapy strategy. Curr Oncol Rep. 2019;21(3):27.
- Dimopoulos MA, Tedeschi A, Trotman J, et al; iNNOVATE Study Group and the European Consortium for Waldenström’s Macroglobulinemia. Phase 3 trial of ibrutinib plus rituximab in Waldenström’s macroglobulinemia. N Engl J Med. 2018;378(25):2399–410.
- Vaxman I, Gertz M. Waldenstrom’s macroglobulinemia in the era of immunotherapy. Leuk Lymphoma. 2020;61(6):1292–304.
- Tam CSL, Opat S, D’Sa S, et al. ASPEN: results of a phase III randomized trial of zanubrutinib versus ibrutinib for patients with Waldenström macroglobulinemia (WM). J Clin Oncol. 2020;38(15_suppl):8007.
- Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: a single-arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112–21.
- Woyach JA, Furman RR, Liu T-M, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–94.
- Woyach JA, Ruppert AS, Guinn D, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(13):1437–43.
- Ahn IE, Underbayev C, Albitar A, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129(11):1469–79.
- Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352–4.
- Jain P, Kanagal-Shamanna R, Zhang S, et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol. 2018;183(4):578–87.
- Jiménez C, Chan GG, Xu L, et al. Genomic evolution of ibrutinib-resistant clones in Waldenström macroglobulinaemia. Br J Haematol. 2020;189(6):1165–70.
- Xu L, Tsakmaklis N, Yang G, et al. Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood. 2017;129(18):2519–25.
- Freedman A, Jacobsen E. Follicular lymphoma: 2020 update on diagnosis and management. Am J Hematol. 2020;95(3):316–27.
- Gopal AK, Kahl BS, Vos S, et al. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008–18.
- Salles G, Schuster SJ, de Vos S, et al. Efficacy and safety of idelalisib in patients with relapsed, rituximab- and alkylating agent-refractory follicular lymphoma: a subgroup analysis of a phase 2 study. Haematologica. 2017;102(4):e156–9.
- Dreyling M, Santoro A, Mollica L, et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol. 2017;35(35):3898–905.
- Dreyling M, Santoro A, Mollica L, et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am J Hematol. 2019. Online ahead of print.
- Flinn IW, Miller CB, Ardeshna KM, et al. DYNAMO: a phase II study of duvelisib (IPI-145) in patients with refractory indolent non-Hodgkin lymphoma. J Clin Oncol. 2019;37(11):912–22.
- Mato AR, Nabhan C, Thompson MC, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874–9.
- O’Brien S, Hillmen P, Coutre S, et al. Safety analysis of four randomized controlled studies of ibrutinib in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma or mantle cell lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(10):648–57.e15.
- Ahn IE. Cardiovascular adverse events of ibrutinib. Blood. 2019;134(22):1881–2.
- Dickerson T, Wiczer T, Waller A, et al. Hypertension and incident cardiovascular events following ibrutinib initiation. Blood. 2019;134(22):1919–28.
- De Weerdt I, Koopmans SM, Kater AP, Van Gelder M. Incidence and management of toxicity associated with ibrutinib and idelalisib: a practical approach. Haematologica. 2017;102(10):1629–39.
- Barr PM, Robak T, Owen C, et al. Sustained efficacy and detailed clinical follow-up of first-line ibrutinib treatment in older patients with chronic lymphocytic leukemia: extended phase 3 results from RESONATE-2. Haematologica. 2018;103(9):1502–10.
- Barrientos JC, O’Brien S, Brown JR, et al. Improvement in parameters of hematologic and immunologic function and patient well-being in the phase III RESONATE study of ibrutinib versus ofatumumab in patients with previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(12):803–13.e7.
- Jain P, Keating MJ, Wierda WG, et al. Long-term follow-up of treatment with ibrutinib and rituximab in patients with high-risk chronic lymphocytic leukemia. Clin Cancer Res. 2017;23(9):2154–8.
- Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015;1(1):80–7.
- Byrd JC, Wierda WG, Schuh A, et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: updated phase 2 results. Blood. 2020;135(15):1204–13.
- Wiczer TE, Levine LB, Brumbaugh J, et al. Cumulative incidence, risk factors, and management of atrial fibrillation in patients receiving ibrutinib. Blood Adv. 2017;1(20):1739–48.
- Khalid S, Yasar S, Khalid A, et al. Management of atrial fibrillation in patients on ibrutinib: a Cleveland Clinic experience. Cureus. 2018;10(5):e2701.
- Brukinsa (zanubrutinib) [prescribing information]. San Mateo, CA: BeiGene USA; 2019.
- Calquence (acalabrutinib) [prescribing information]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2019.
- Imbruvica (ibrutinib) [prescribing information]. Sunnyvale, CA: Pharmacyclics LLC; and Horsham, PA: Janssen Biotech, Inc.; 2020.
- Bye AP, Unsworth AJ, Desborough MJ, et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1(26):2610–23.
- Kunk PR, Mock J, Devitt ME, et al. Major bleeding with ibrutinib: more than expected. Blood. 2016;128(22):3229.
- Serebruany VL, Malinin AI, Ferguson JJ, et al. Bleeding risks of combination vs. single antiplatelet therapy: a meta-analysis of 18 randomized trials comprising 129,314 patients. Fundam Clin Pharmacol. 2008;22(3):315–21.
- Awan FT, Schuh A, Brown JR, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019;3(9):1553–62.
- Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125(16):2497–506.
- Howard SC, Jones DP, Pui C-H. The tumor lysis syndrome. N Engl J Med. 2011;364(19):1844–54.
- Venclexta (venetoclax) [prescribing information]. North Chicago, IL: AbbVie Inc.; and South San Francisco, CA: Genentech USA; 2019.
- Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107–20.
- Verma A, Mbughuni M, Mariash E, Mesa H. Hyperleukocytosis increases risk of fatal hyperkalemia with new ibrutinib/venetoclax regimen for refractory mantle cell lymphoma. J Chemother. 2019;31(7-8):428–31.
- Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768–78.
- Zydelig (idelalisib) [prescribing information]. Foster City, CA: Gilead Sciences, Inc.; 2018.
- Copiktra (duvelisib) [prescribing information]. Needham, MA: Verastem, Inc.; 2018.
- Aliqopa (copanlisib) [prescribing information]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc.; 2017.
- Coutré SE, Barrientos JC, Brown JR, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;56(10):2779–86.
- O’Brien SM, Lamanna N, Kipps TJ, et al. A phase 2 study of idelalisib plus rituximab in treatment-naïve older patients with chronic lymphocytic leukemia. Blood. 2015;126(25):2686–94.
- Morschhauser F, Machiels JP, Salles G, et al. On-target pharmacodynamic activity of the PI3K inhibitor copanlisib in paired biopsies from patients with malignant lymphoma and advanced solid tumors. Mol Cancer Ther. 2020;19(2):468–78.
- Patnaik A, Appleman L, Tolcher A, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016;27(10):1928–40.
- Cheson BD, O’Brien S, Ewer MS, et al. Optimal management of adverse events from copanlisib in the treatment of patients with non-Hodgkin lymphomas. Clin Lymphoma Myeloma Leuk. 2019;19(3):135–41.
- Dreyling M, Morschhauser F, Bouabdallah K, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28(9):2169–78.
Back to Top