
Expired activity
Please go to the PowerPak
homepage and select a course.
New Directions in the Management of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: Utilizing Immunotherapy to Improve Outcomes
Current and Emerging First-line Immune Checkpoint Inhibitor Treatment Options in Recurrent or Metastatic HNSCC
Immunotherapy MOA
Immune checkpoints – cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) and PD-1/PD-L1/PD-L2 – negatively regulate T-cell immune function, and their inhibition results in increased activation of the immune system, which has revolutionized treatment options for patients with various cancer types. Cellular immunity results from tumor antigen recognition by antigen presenting cells (APCs), leading to immune activation through interactions of B7.1/B7.2
and CD28 on the cell surface of the APCs and the resting T cell.1 In contrast, CTLA-4, often upregulated on T cells after antigen exposure, competes with CD28 with much higher affinity for binding to B7.1/B7.2, resulting in a negative signal for early T-cell activation. Similarly, PD-L1 binds to PD-1, a member of the B7/CD28 family of stimulatory receptors, leading to immune inactivation mainly at tumor sites. The anticipation about using immunotherapy to treat head and neck cancers has been driven primarily by results from clinical studies evaluating antagonist antibodies to PD-1 and PD-L1, which have demonstrated prolonged tumor responses in patients with advanced disease.
Because CTLA-4 and PD-1 operate at different stages of the immune pathways, differences can be observed with regard to efficacy, toxicity, and adverse effects. Inhibition of CTLA-4, which results earlier in the immune cascade, is believed to activate a larger number of T cells, resulting in more effective antitumor responses and depletion or inhibition of regulatory T cells in tumors.2,3 Inhibition of CTLA-4 may also lead to activation of T cells specific to a wide range of antigens, thus explaining the increased immune-mediated adverse drug reactions (ADRs) observed with this class of drugs. In comparison, PD-1 inhibition occurs primarily at the site of the tumor during the effector phase, predominantly within peripheral tissues, resulting in a more restricted spectrum of T-cell activation.1 These mechanistic differences are likely the reasons for the decreased incidence of immune-mediated ADRs seen with PD-1/PD-L1 inhibitors.4 Furthermore, it is believed that inhibiting PD-L1 provides an even more-targeted signal with less unwanted toxicity, as self-tolerance mediated through PD-L2 interactions with PD-1 are preserved.4,5 However, it is hypothesized that targeting the ligand may not be as efficacious as targeting the receptor.
Despite the clinical benefit that checkpoint inhibitors have had, immune-mediated ADRs remain a major topic of interest. These ADRs range from dermatologic, gastrointestinal, hepatic, and endocrine to other inflammatory events and are thought to arise from immune stimulation.6 In general, management involves withholding treatment for grade 1/2 toxicities and administering corticosteroids with supportive care. Grade 3/4 ADRs typically require permanent discontinuation of treatment, administration of corticosteroids, and administration of antitumor necrosis factor agents in life-threatening or refractory cases.
Nivolumab and pembrolizumab are both monoclonal antibodies that bind to PD-1 receptors and block their interaction with PD-L1 and PD-L2, which releases PD-1 pathway-mediated inhibition of the immune response, including the antitumor immune response.7,8 The blocking of PD-1 activity might result in decreased tumor growth. The overall response rate to nivolumab and pembrolizumab has been much lower in patients with HNSCC than in those with Hodgkin’s lymphoma (15% vs 87%).9 An estimated 50% of patients with HNSCC treated with PD-1/PDL-1 inhibitors experience treatment-related adverse events that negatively affect treatment outcomes. Additional research is warranted to understand the pattern and mechanisms of PD-1/PD-L1 expression in HNSCC.
Efficacy and Safety Data in First-Line Head and Neck Cancers
For patients with metastatic (M1) disease at initial presentation, palliative adjunctive measures include radiotherapy (RT), surgery, analgesics, and other therapies to control manifestations of disease spread (eg, pain, hypercalcemia, malnutrition). Locoregional treatment (eg, surgery, RT, or ablative therapies) may be used for oligometastatic disease.10-12 Historically, single-agent and combination systemic therapy have both been used.13 Response rates to single-agent therapies range from 15% to 35%.14-16 Randomized trials assessing a cisplatin-based combination regimen (cisplatin/5-FU) vs single-agent therapy with cisplatin, 5-FU, or methotrexate showed significantly higher response rates, but no difference in overall survival (OS) and greater toxicity for the combination regimen.17-21 Complete response is associated with longer survival and, although infrequent, has been reported more often with combination regimens.18
Trials evaluating immune checkpoint inhibitors demonstrated efficacy in patients with recurrent or metastatic HNSCC.22-24 Pembrolizumab was evaluated as a first-line option for recurrent or metastatic HNSCC in the KEYNOTE-048 trial (N = 882).22 Patients were randomized to receive pembrolizumab, pembrolizumab with a platinum and 5-FU, or the EXTREME regimen. In the total population, an OS benefit was observed in the pembrolizumab/platinum/5-FU arm, compared to the EXTREME arm (median OS 13 months vs 10.7 months, respectively; HR, 0.77; 95% CI, 0.63–0.93; P = 0.003). Progression-free survival (PFS), however, did not significantly differ between these two study arms. In patients with a PD-L1 combined positive score (CPS) of ≥ 20 or ≥ 1, median OS was better in patients who received pembrolizumab monotherapy, compared to those who received the EXTREME regimen (median 14.9 months vs 10.7 months, respectively; HR, 0.61; 95% CI, 0.45–0.83; P < 0.001, for CPS ≥ 20; median 12.3 months vs 10.3 months, respectively; HR, 0.78; 95% CI, 0.64–0.96; P = 0.009, for CPS ≥ 1). Median duration of response was greater in patients treated with pembrolizumab monotherapy or pembrolizumab with chemotherapy, compared to patients treated with the EXTREME regimen. It should be noted that grade 3–5 toxicity was observed in 85% of patients receiving pembrolizumab/platinum/5-FU, and in 55% of patients on pembrolizumab monotherapy.
The NCCN panel considers immunotherapy as the preferred first-line systemic therapy option for all patients with recurrent, unresectable, or metastatic disease who have no surgical or radiotherapeutic option.25 Specifically, pembrolizumab/platinum/5-FU is a category 1 option based on the results of KEYNOTE-048, and this combination regimen may be particularly suitable in patients with a PS of 0 or 1 and either a large burden of disease or nearing a clinical crisis. The NCCN panel also considers pembrolizumab monotherapy to be a preferred first-line option for patients with CPS ≥ 1 (category 1 if CPS ≥ 20). A taxane can be substituted for 5-FU, when used in combination with pembrolizumab and a platinum, but this is a category 2B option based on less panel consensus and evidence. Single agents recommended by the panel include cisplatin, carboplatin, paclitaxel, docetaxel, 5-FU, methotrexate, capecitabine, and cetuximab.
Current and emerging subsequent-line immune checkpoint inhibitor treatment options in recurrent or metastatic HNSCC
Nivolumab was assessed in a phase 3 RCT, CheckMate 141, including 361 patients with recurrent HNSCC whose disease had progressed within 6 months following platinum-based chemotherapy.24,26 With a median follow-up of 5.1 (range 0–16.8) months, the OS was significantly greater in patients given nivolumab, compared to patients given standard second-line single-agent systemic therapy (methotrexate, docetaxel, or cetuximab), (HR, 0.70; 97.73% CI, 0.51–0.96; P = 0.01). One-year survival was also greater for patients who received nivolumab, relative to patients who received standard therapy (36.0% vs 16.6%, respectively), and response rate was higher (13.3% vs 5.8%, respectively), but median PFS was not significantly different between the two groups (2.0 months vs 2.3 months, respectively; P = 0.32). In prespecified exploratory analyses, the OS benefit in patients treated with nivolumab appeared to be confined to those patients with a tumor PD-L1 expression level of 1% or more (n = 149) (8.7 vs 4.6 months, HR, 0.55; 95% CI, 0.36–0.83). In patients with tumor PD-L1 expression level less than 1% (n = 111), no OS advantage was demonstrated for the nivolumab-treated patients (5.7 vs 5.8 months; HR, 0.89; 95% CI, 0.54–1.45). Grade 3 or 4 treatment-related adverse events occurred in 13.1% of patients who received nivolumab, compared to 35.1% of patients who received standard therapy. These results indicate that nivolumab prolongs survival in patients with recurrent or metastatic squamous cell H&N cancer that has progressed after platinum-based chemotherapy, relative to patients who receive standard single-agent systemic therapy. There are two FDA-approved dosing regimens for nivolumab for treatment of HNSCC: 240 mg every 2 weeks or 480 mg ever 4 weeks.7
Pembrolizumab was initially studied at a dose of 10 mg/kg given every 2 weeks in the HNSCC cohort of the KEYNOTE-012 trial, and clinical activity was identified.27 A lower, fixed-dose schedule using pembrolizumab 200 mg every 3 weeks was subsequently assessed in a phase 1b expansion cohort of 132 patients with recurrent or metastatic HNSCC.28 At 6 months, the OS rate was 59%, and the PFS was 23%, with an ORR of 18%. Observed responses appeared durable, although the follow-up was limited (median 9 months). Pembrolizumab was also generally well-tolerated.27 Pooled analyses after long-term follow-up of the initial and expansion cohorts (N = 192) showed a 1-year OS rate of 38%.29 Among the 34 responders, 85% of the responses lasted 6 months or longer, and 71% lasted 12 months or longer. The FDA has approved an alternate dosing regimen of pembrolizumab 400 mg every 6 weeks across all currently approved adult indications.8 Based on results of the phase 1b KEYNOTE-012 trial, pembrolizumab was evaluated in the phase 3 KEYNOTE-040 trial.30 Patients with recurrent or metastatic HNSCC (N = 495) were randomized to receive pembrolizumab or another systemic therapy (methotrexate, docetaxel, or cetuximab). Median OS was greater for the pembrolizumab arm compared to the standard-of-care arm (8.4 months vs 6.9 months; HR, 0.80; 95% CI, 0.65–0.98; P = 0.016). When analyses were stratified by PD-L1 status, the results for OS were significantly better with pembrolizumab only for patients with tumors that have PD-L1 expression. Pembrolizumab monotherapy was also evaluated for previously treated tumors with high microsatellite instability (MSI-H) in the phase 2 KEYNOTE-158 basket trial, which included 1 patient with HNSCC.31 The ORR for the entire sample (N = 233) was 34.3% (95% CI, 28.3%–40.8%), median PFS was 4.1 months (95% CI, 2.4–4.9), and median OS was 23.5 months (95% CI, 13.5 months–not reached). The nonrandomized phase 2 KEYNOTE-055 trial studied pembrolizumab in 171 patients with HNSCC that progressed following treatment with both a platinum and cetuximab.32 The ORR was 16% (95% CI, 11%–23%), and the mean duration of response was 8 months.
The NCCN panel recommends immunotherapy (nivolumab and pembrolizumab) as category 1 preferred options for patients with recurrent or metastatic HNSCC who have progressed on or following platinum-based chemotherapy based on high-quality evidence.25 Pembrolizumab is also an option for treatment of MSI-H disease. Despite the ambiguities of PD-L1 testing and definitions, PD-L1 expression may be associated with better outcomes from treatment with immunotherapy for recurrent or metastatic HNSCC (ie, greater likelihood of response to pembrolizumab and greater survival benefit in response to nivolumab).
Pharmacist Considerations in Patient Management
As a class, the spectrum of adverse effects associated with immune checkpoint inhibitors is very different than other systemic anticancer agents. irAEs commonly affect skin, GI tract, lungs, and endocrine, thyroid, adrenal, pituitary, musculoskeletal, renal, nervous, hematologic, cardiovascular, and ocular systems.33,34 Due to the unpredictable nature of irAEs, there should be a high level of suspicion that any changes in the patient’s symptoms or quality of life are treatment-related.
A major practical difference in AEs associated with chemotherapy vs those associated with immunotherapy is that traditional cytotoxic chemotherapy often results in acute-onset emetic and myelosuppressive effects, while irAEs tend to be relatively delayed-onset and inflammatory or autoimmune in nature. Accordingly, close follow-up of patients and timely management is critical to minimize morbidity. In each case, the health care provider is charged with determining answers to a set of basic clinical decisions including: (1) assessing whether it is autoimmune etiology or other cause; (2) whether to hold or continue treatment; (3) the timing of steroid initiation; (4) the starting dose of steroid and duration of treatment; (5) the route of administration of steroids (oral vs IV); (6) whether it can be handled as an inpatient vs outpatient; and (7) when to initiate second-line immunosuppressive therapy.35
| Figure. Onset of Immune Checkpoint Inhibitor-Associated Toxicities37-39 |
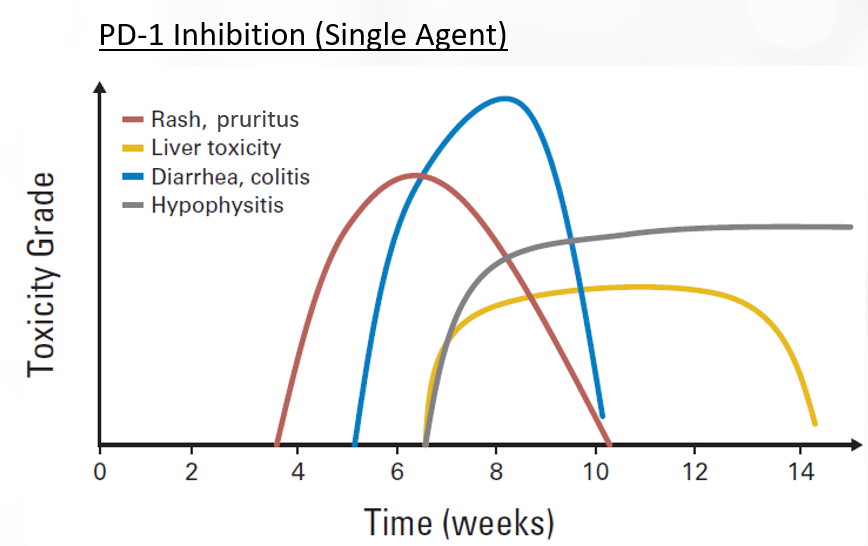 |
irAEs are typically inflammatory or autoimmune in nature.36-38 There is a trend toward higher incidence of irAEs when these agents are used in combination therapy (ie, dual immune checkpoint inhibitor, targeted, and chemotherapies).33,36 Toxicities affect all organ systems, including the GI tract (colitis, diarrhea), lung (pneumonitis), endocrine system (hypophysitis, thyroiditis), liver (hepatitis) and skin (rash, pruritus), among others. irAEs do not necessarily appear concurrently. For instance, skin and GI events usually appear within 1 to 2 cycles of dual blockade, while hepatitis, pneumonitis, and endocrine side effects appear later.
There is concern that the administration of immune-activating agents may exacerbate pre-existing autoimmune disease.33 Data on the toxicity of immune checkpoint inhibitors in patients with preexisting autoimmune disease or prior irAEs are generally lacking due to exclusion of these populations from the registry clinical trials leading to FDA approval. However, based on limited data from smaller retrospective studies, immune checkpoint inhibitors appear to have a similar efficacy in these patient groups, as in the general population. A retrospective records analysis of patients with NSCLC and a history of an autoimmune disorder (AID) found that checkpoint inhibitor therapy did not significantly aggravate the AID for most patients and that irAE rates were similar to those reported in clinical trials that excluded AIDs.39
Close consultation with disease-specific subspecialists is encouraged during irAE management.33 Routine premedication with corticosteroids is not recommended.
- Infusion-related reactions (IRRs) are uncommon and mild. IRRs are associated with low-grade fever, chills, headache, or nausea. Mild IRRs do not require infusion interruption or other interventions. For moderate IRRs, the infusion rate should be slowed or held, and antihistamines, acetaminophen, NSAIDS, narcotics, or IV fluids may be required. Severe IRRs are often more prolonged with limited responsiveness to intervention or infusion interruption and can recur following initial improvement.
- Dermatological toxicities are the most common irAE and typically present within the first 2 cycles of therapy. Maculopapular rash, pruritus, vitiligo, alopecia, hair repigmentation, and eczematous, lichenoid, psoriasiform, and bullous dermatitis have been observed. The reported incidences of all grade dermatologic toxicity range from 37% to 70% for ipilimumab and 17% to 40% for PD-1/PD-L1 inhibitors, with a similar 1% to 3% of patients experiencing high-grade dermatological toxicity across all immune checkpoint inhibitors. In general, short-term use of higher potency topical corticosteroids is preferred over longer-term use of a lower-potency agent.
- Gastrointestinal toxicity may present as diarrhea or colitis, typically 6 to 8 weeks after therapy initiation. The incidence is highest in patients receiving dual nivolumab/ipilimumab therapy, high in patients receiving anti-CTLA-4 therapy and least common in patients receiving anti-PD-L1 and anti-PD-1 therapies (13.6%, 9.1%, 1.3%, respectively). Corticosteroids will resolve GI toxicity in 40% to 60% of patients.
- Hepatic toxicity is rare, typically mild, but can be fatal. The incidence is estimated at 3% to 9% for ipilimumab and 0.7% to 1.8% for anti-PD-L1 and anti-PD-1 therapies. Autoimmune hepatitis and drug-induced hepatitis can be difficult to distinguish but may be differentiated by distinct histologic features and imaging.
- Pancreatic toxicity is common and associated with elevated amylase and/or lipase levels, but do not typically require intervention. Acute pancreatitis is rare and treated with standard medical care, including hospital admission, aggressive fluid resuscitation, and pain control. Gastroenterology consultation and immunosuppression are warranted if clinical assessment and/or imaging findings support moderate/severe acute pancreatitis.
- Endocrine dysfunction typically involves the thyroid, pituitary, adrenal glands, and pancreas and is associated with hypothyroidism, hyperthyroidism, hypophysitis, type I diabetes, and primary adrenal insufficiency. Endocrine toxicity is very challenging to distinguish from other treatment- and disease-related causes. Median time to onset of moderate-to-severe endocrinopathy has ranged from 1.75 to 5 months for ipilimumab and 1.4 to 4.9 months with PD-1 inhibitor monotherapy. Life-long hormone replacement therapy may be required for some patients.
- Pneumonitis is rare (< 5% all-grade, ≥ 1% high-grade) but may be fatal. The median time to irAE onset has been reported at 2.5 months (earlier for combination vs monotherapy).
- Nervous system toxicity is moderately common (3.8% for CTLA-4 inhibitors, 6% with PD-1 inhibitors, and 12% for combination therapy) and may be fatal, despite aggressive treatment. Neurologic irAEs include numerous conditions such as myasthenia gravis, Guillain-Barré Syndrome (GBS)-like syndrome, central and/or peripheral neuropathy, aseptic meningitis, encephalitis, and transverse myelitis. Fatalities were more commonly associated with encephalitis and myasthenia gravis. Prompt treatment is critical for reducing long-term morbidity and mortality. Resolution is typically < 8 weeks with treatment.
- Cardiovascular toxicities, including myocarditis, cardiomyopathy, cardiac fibrosis, heart failure, and cardiac arrest are uncommon but may be fatal. Prevalence has been estimated to be 1.1%, but may be underestimated, and with a median onset of 34 days from initiation of treatment. Patients with suspected cardiotoxicity should have a full cardiac work up and receive corticosteroids until cardiac function returns to normal, with a 4- to 6-week taper.
- Musculoskeletal toxicities include inflammatory arthritis, myositis, and myalgias. Myositis can be fatal and is more commonly observed in patients receiving anti-PD-1/PD-L1 therapy than anti-CTLA-4 therapy. The incidence is up to 7%.
The NCCN, American Society of Clinical Oncology (ASCO), and European Society for Medical Oncology (ESMO) released a clinical practice guideline for the management of irAEs in patients treated with immune checkpoint inhibitor therapy.33,34,40 These organizations recognized the need for greater awareness and guidance on the management of irAEs. They have provided grading and management. The recommendations include:
- Patient and family caregivers should receive education about immunotherapies and irAEs prior to initiating therapy and throughout treatment and survivorship.
- There should be a high level of suspicion that new symptoms are treatment related.
- In general, therapy should be continued with close monitoring for grade 1 toxicities, with the exception of some neurologic, hematologic, and cardiac toxicities.
- Hold treatment for most grade 2 toxicities; resume when symptoms revert to grade 1 or less. Corticosteroids may be administered.
- Hold for grade 3 toxicities and initiate high-dose corticosteroids. Taper corticosteroids over 4 to 6 weeks. Infliximab (anti-TNFα) may be offered if symptoms do not improve in 2 to 3 days. Use caution when resuming after resolution of symptoms to grade ≤ 1; do not adjust dose.
- Permanent discontinuation for grade 4 toxicities, except for select endocrinopathies (ie, hypothyroidism controlled with levothyroxine supplementation).
| Table. Management of Immune-Related Toxicities Associated with Checkpoint Inhibitors41 |
| irAE |
ICI Therapy |
Immunosuppressants |
Other Treatment |
| Grade 1 |
Discontinue if hypophysitis, pneumonitis, and/or sarcoidosis. Consider holding if renal. Hold if neurologic, aplastic anemia, acquired hemophilia. Continue for all others |
Prednisone 0.5-1 mg/kg/d if acquired hemophilia |
Topical steroids , oral antihistamines , topical emollients if dermatologic |
| Loperamide if gastrointestinal |
| Thyroid hormone supplementation if hypothyroidism |
| Beta-blockers for symptomatic hyperthyroidism; insulin therapy if hyperglycemia |
| Oral fluids, loperamide, hormone replacement therapy if hypophysitis |
| Consider artificial tears if ocular |
| Analgesics if rheumatologic |
| Grade 2 |
Considering holding if dermatologic, rheumatologic, or lymphopenia. Hold for all others |
Prednisone 0.5-1 mg/kg/d
Prednisone 1-2 mg/kg/d if hypophysitis
Prednisone 2 mg/kg/d if transverse myelitis |
In addition to the above, consider: adding infliximab if gastrointestinal |
| Empiric antibiotics if pulmonary |
| Prednisone 2 mg/kg/d if transverse myelitis |
| Adding ATG and cyclosporine if aplastic anemia |
| Adding GABA agonist l or duloxetine for pain if peripheral neuropathyAdding ophthalmic prednisone if ocular |
| Grade 3 |
Discontinue if hepatitis, renal, ocular, neurologic, cardiovascular, rheumatologic, and/or hematologic. Hold for all others |
Prednisone 1-2 mg/kg/d Prednisone 2-4 mg/kg/d if peripheral neuropathy or Guillain-Barré syndrome. Consider plasmapheresis, intravenous immunoglobulin therapy, methotrexate, azathioprine, or mycophenolate mofetil through grade 4 if myositis;
Consider rituximab or cyclophosphamide if acquired hemophilia |
In addition to the above, consider: adding omalizumab, GABA agonist if pruritis. Plasmapheresis or immunoglobulin if neurologic. Pyridostigmine if myasthenia gravis. Antirheumatic drugs, methotrexate, infliximab, or tocilizumab if refractory arthritis or polymyalgia-like syndrome. Infliximab, mycophenolate mofetil, intravenous immunoglobulin if pulmonary or renal Rituximab if autoimmune encephalopathy. Infliximab if cardiovascular |
| Grade 4 |
Discontinue |
Prednisone 2-4 mg/kg/d |
In addition to the above, consider: adding mycophenolate mofetil if hepatitis
Empiric antivirals if aseptic meningitis and/or encephalitis. Rituximab if acquired TTP.
Rituximab or cyclophosphamide if acquired hemophilia rituximab, intravenous immunoglobulin, cyclosporine A, or mycophenolate mofetil if autoimmune hemolytic anemia. Eculizumab if hemolytic uremic syndrome.
Intravenous immunoglobulin, rituximab, or thrombopoietin receptor agonists if immune thrombocytopenia |
References
- Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Ann Rev Immunol. 2008;26:677-704.
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717-1725.
- Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 anti-bodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32-42.
- Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300-5309.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264.
- Champiat S, Lambotte O, Barreau E, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2016;27:559.
- Prescribing information. Bristol Myers Squibb; 2022. Accessed April 28, 2022. https://packageinserts.bms.com/pi/pi_opdivo.pdf.
- Prescribing information. Merck & Co, Inc; 2022. Accessed April 28, 2022. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf.
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576-582. doi:10.1038/nature14129.
- Sun XS, Michel C, Babin E, et al. Approach to oligometastatic disease in head and neck cancer, on behalf of the GORTEC. Future Oncol. 2018;14:877-889.
- Bonomo P, Greto D, Desideri I, et al. Clinical outcome of stereotactic body radiotherapy for lung-only oligometastatic head and neck squamous cell carcinoma: is the deferral of systemic therapy a potential goal? Oral Oncol. 2019;93:1-7.
- Bates JE, De Leo AN, Morris CG, et al. Oligometastatic squamous cell carcinoma of the head and neck treated with stereotactic body ablative radiotherapy: single-institution outcomes. Head Neck. 2019;41:2309-2314.
- Fury MG, Pfister DG. Current recommendations for systemic therapy of recurrent and/or metastatic head and neck squamous cell cancer. J Natl Compr Cancer Netw. 2011;9:681-689.
- Price KA, Cohen EE. Current treatment options for metastatic head and neck cancer. Curr Treat Options Oncol. 2012;13:35-46.
- Molin Y, Fayette J. Current chemotherapies for recurrent/metastatic head and neck cancer. Anticancer Drugs. 2011;22:621-625.
- Hoffmann TK. Systemic therapy strategies for head-neck carcinomas: current status. GMS Curr Top Otorhinolaryngol Head Neck Surg. 2012;11:Doc03.
- Gibson MK, Li Y, Murphy B, et al. Randomized phase III evaluation of cisplatin plus fluorouracil versus cisplatin plus paclitaxel in advanced head and neck cancer (E1395): an intergroup trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2005;23:3562-3567.
- Forastiere AA, Metch B, Schuller DE, et al. Randomized comparison of cisplatin plus fluorouracil and carboplatin plus fluorouracil versus methotrexate in advanced squamous-cell carcinoma of the head and neck: a Southwest Oncology Group study. J Clin Oncol. 1992;10:1245-1251.
- Jacobs C, Lyman G, Velez-Garcia E, et al. A phase III randomized study comparing cisplatin and fluorouracil as single agents and in combination for advanced squamous cell carcinoma of the head and neck. J Clin Oncol. 1992;10:257-263.
- Browman GP, Cronin L. Standard chemotherapy in squamous cell head and neck cancer: what we have learned from randomized trials. Semin Oncol. 1994;21:311-319.
- Clavel M, Vermorken JB, Cognetti F, et al. Randomized comparison of cisplatin, methotrexate, bleomycin and vincristine (CABO) versus cisplatin and 5-fluorouracil (CF) versus cisplatin (C) in recurrent or metastatic squamous cell carcinoma of the head and neck. A phase III study of the EORTC Head and Neck Cancer Cooperative Group. Ann Oncol. 1994;5:521-526.
- Burtness B, Harrington KJ, Greil R, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE- 048): a randomised, open-label, phase 3 study. Lancet. 2019;394:1915-1928.
- Cohen EEW, Soulieres D, Le Tourneau C, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393:156-167.
- Ferris RL, Blumenschein G, Jr., Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856-1867.
- The NCCN Clinical Practical Guidelines in Oncology Head and Neck Cancers (Version 1.2022); 2021; www.NCCN.org. Available from: https://www.nccn.org/professionals/physician_gls/pdf/head-and-neck.pdf. Accessed April 28, 2022.
- Gillison ML, Blumenschein G, Fayette J, et al. Checkmate 141: 1-year update and subgroup analysis of nivolumab as first-line therapy in patients with recurrent/metastatic head and neck cancer. Oncologist. 2018;23(9):1079-1082.
- Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956-965.
- Chow LQ, Haddad R, Gupta S, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. J Clin Oncol. 2016;34:3838-3845.
- Mehra R, Seiwert TY, Gupta S, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: pooled analyses after long-term follow-up in KEYNOTE-012. Br J Cancer. 2018;119:153-159.
- Cohen EEW, Soulieres D, Le Tourneau C, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393:156-167.
- Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. 2020;38:1-10.
- Bauml J, Seiwert TY, Pfister DG, et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J Clin Oncol. 2017;35:1542-1549.
- National Comprehensive Cancer Network. NCCN Guidelines Version 1.2022, Management of Immunotherapy-Related Toxicities. https://www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf.
- Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018;36(17):1714-1768.
- Sznol M. Immunotherapy for melanoma: management of adverse events. Personal Communication and Presentation at: PER’s 11th Annual International Symposium on Melanoma and Other Cutaneous Malignancies. March 7, 2015.
- Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy. 2016;8(7):821-837.
- Marrone KA, Ying W, Naidoo J. Immune-related adverse events from immune checkpoint inhibitors. Clin Pharmacol Ther. 2016;100(3):242-251.
- Postow MA, Sidlow R, Hellmann M. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158-168.
- Leonardi GC, Gainor JF, Altan M, et al. Safety of programmed death-1 pathway inhibitors among patients with non-small cell lung cancer and preexisting autoimmune disorders. J Clin Oncol. 2018;36(19):1905-1912.
- Haanen J, Carbonnel F, Robert C, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines. Ann Oncol. 2017;28(suppl 4):iv119-iv142.
- Trinh S, Le A, Gowani S, La-Beck NM. Management of immune-related adverse events associated with immune checkpoint inhibitor therapy: a minireview of current clinical guidelines. Asia-Pacific Journal of Oncology Nursing. 2019;6(2):154-160.
Back to Top