
Expired activity
Please go to the PowerPak
homepage and select a course.
COVID-19 Quarterly Update: Omicron Persists — Implications for Vaccination and Treatment
INTRODUCTION
As the story of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 2019 (COVID-19) enters its fourth year, many people have “moved on” from the pandemic. The virus has seemingly become less virulent for many people, and the efficacy of currently available vaccines against circulating omicron subvariants is questionable. A case of COVID-19 in a school or workplace is a “nothing burger” — the ill person stays home and everyone else goes on with their daily lives. President Biden has announced that the COVID-19 national and public health pandemic emergency declarations are ending on May 11, 2023.
The numbers back up this view of COVID-19. Cases and deaths in the United States have plateaued, as shown in Figure 1. In the week ending on February 1, 2023, the 280,911 new cases reported to the U.S. Centers for Disease Control and Prevention (CDC) were at their lowest point since the respite in the summer of 2021. The number of deaths that week (3,452) was likewise low, with no dramatic winter surge in morbidity and mortality (Figure 2). Only 343,044 vaccine doses were administered that week, and less than 16% of Americans were vaccinated with the updated bivalent booster, according to the CDC.
What some might call a sense of apathy among Americans is concerning. The low uptake of the bivalent vaccination could translate into increased morbidity and mortality of disease, particularly within the most vulnerable populations, such as older adults and people with immunocompromising conditions. When disease occurs, safe and effective treatments are not widely used to prevent ambulatory patients with mild-to-moderate conditions from progressing to severe COVID-19. Treatments to decrease mortality in hospitals are limited in number and are expensive.
This PowerPak continuing pharmacy education program provides a quarterly update for frontline pharmacists and pharmacy technicians of information needed for answering the questions of patients and professional colleagues and providing state-of-the-art care for preventing and treating COVID-19.
PREVENTION OF COVID-19: VACCINES AND MEDICATIONS VS. A(NOTHER) NEW SUBVARIANT
One of the big surprises during the COVID-19 pandemic has been the frequency and rapidity of mutations that enable SARS-CoV-2 to evade inactivation by vaccine- or disease-induced antibodies and the protective effects of pharmaceutical preparations.
Since PowerPak’s last quarterly COVID-19update in November 2022, the predominant SARS-CoV-2 omicron subvariant has shifted from BA.5 to BQ.1 and B1.1 and now to XBB.1.5. When this program was prepared in February 2022, the XBB.1.5 strain had taken over the COVID-19 landscape in the United States, accounting for nearly three-fourths of strains identified by the CDC for the week ending on February 11, 2023, followed by BQ.1.1 (15.3%) and BA.1 (5.1%). In the northeastern United States, an increase in hospitalizations in New York State due to COVID-19 was driven by the emergence of XBB.1.5 and other factors, including increased interactions of people during holiday gatherings.
Bivalent COVID-19 Vaccine Authorized for Nearly Everyone, But Uptake Is Low
The bivalent COVID-19 vaccines authorized by the U.S. Food and Drug Administration (FDA) in late August 2022 contain mRNA from the original vaccine parent strain and an mRNA component in between the omicron BA.4 and BA.5 lineages that were causing the most disease in fall 2022. Both the Pfizer/BioNTech bivalent vaccine and the Moderna bivalent option are now authorized for infants starting at 6 months of age and all children, adolescents, and adults (Table 1).
With this authorization, the third dose of the Pfizer monovalent primary series is no longer authorized in this age group, as the bivalent vaccine takes its place as the final dose. Safety and efficacy data to support this recommendation were derived from earlier data on the monovalent primary series in children of this age group, as well as the immune response of the investigational Pfizer-BioNTech bivalent vaccine as a second booster in adults over the age of 55 years. Children 6 months to 4 years of age who have already received the third dose of the monovalent vaccine are not eligible to receive the bivalent vaccine at this time, but the FDA anticipates receiving data to support this use.
In another development, the FDA Vaccines and Related Products Advisory Committee (VRBPAC) voted to move toward a more unified strain composition for both COVID-19 primary and booster vaccines for those patients who have not yet been vaccinated. Members unanimously support using a bivalent formulation for the primary and booster doses of COVID-19 vaccine based on contemporaneous trends in variant and subvariant infections. This vote aligns with the European Medicines Agency Emergency Task Force who recommended similar simplification in June 2022.
The VRBPAC vote occurred after data from multiple regulatory organizations — including the CDC, FDA, and the National Institutes of Health (NIH) —were presented that demonstrate fewer hospitalizations and deaths in patients receiving bivalent boosters. However, despite such trends, overall uptake of the bivalent vaccines has been poor, with approximately 16% of all eligible populations receiving an updated booster bivalent vaccine. Rates of receipt of the bivalent mRNA vaccine have been 19.2% in the 18 and older population and 39% in those 65 years of age or older.
What factors are causing this low adoption rate for the bivalent vaccine products? Several aspects of this conundrum deserve discussion.
First, clinical outcome data regarding BQ.1, BQ.1.1, and now importantly XBB.1.5 strains are limited. Several studies have shown benefits from the bivalent vaccines in the current environment, but the rapid changes in the predominant strains make their applicability uncertain. In data published by the CDC, patients who received the updated bivalent vaccine were much less likely to be hospitalized from COVID-19 than comparator groups (Figure 3). Compared with individuals vaccinated with the bivalent booster, the hospitalization rate was 16 times higher in unvaccinated individuals and 2.6 times higher in those vaccinated only with the monovalent products. Individuals who had received a primary series but no boosters had some benefit, but much less than those receiving 1 or 2 booster doses.
| Figure 3. Age-Adjusted Rates of COVID-19-Associated Hospitalizations Among Adult Americans by Vaccination Status, January 2021 to November 2022 |
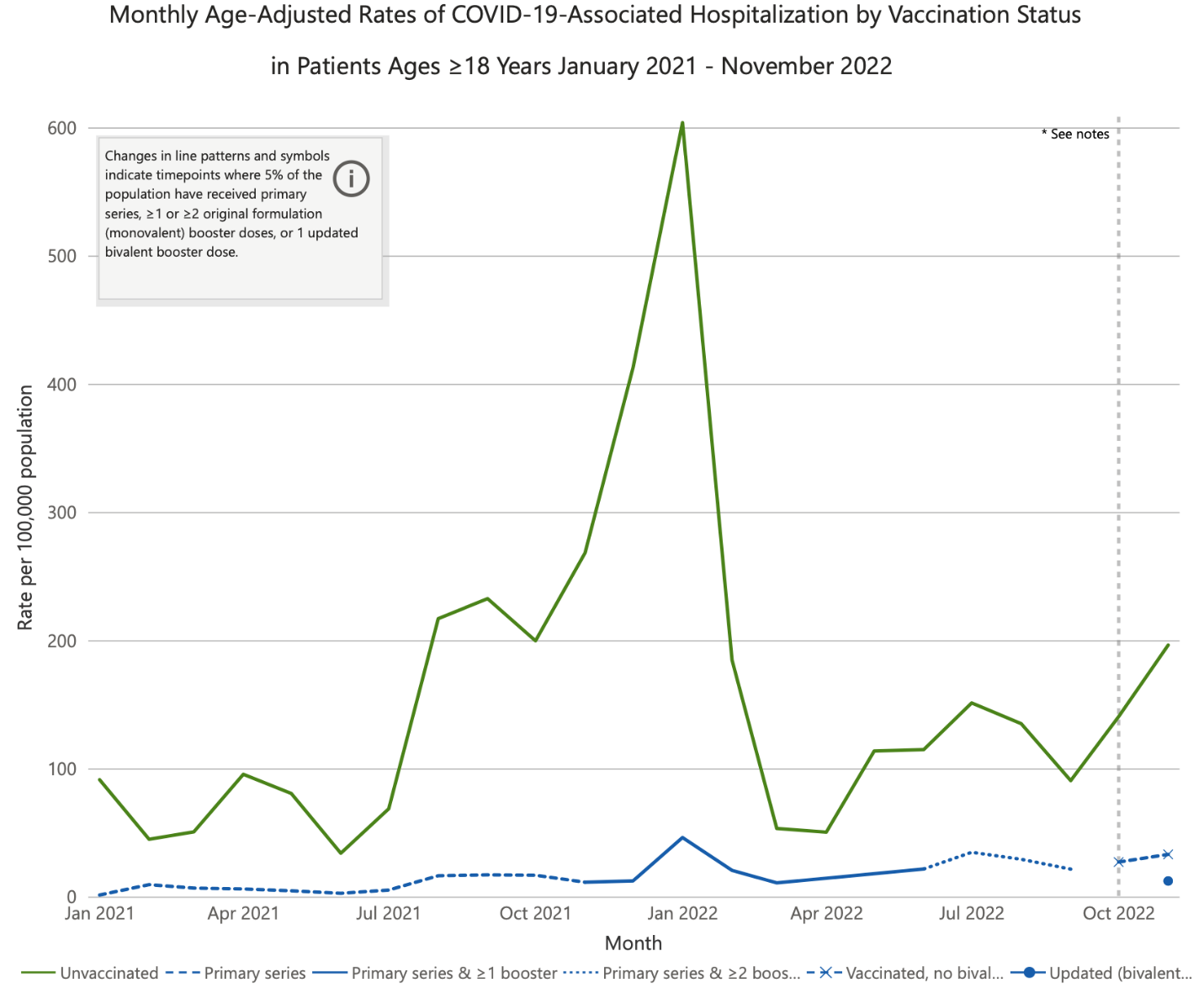 |
Source: Centers for Disease Control and Prevention.
Data were posted on December 28, 2022, and reflect hospitalizations through November 2022. |
Other reports have presented mixed evidence for the bivalent vaccine. In the New England Journal of Medicine, a study showed minimal additional neutralizing antibodies with bivalent doses over original monovalent boosters.1 Concern over the lack of benefits, including “immunologic imprinting,” was discussed in an opinion piece also published in the New England Journal of Medicine by Paul A. Offit, M.D., a member of the VRBPAC and a respected virologist and vaccinologist. He wrote2:
“Why did the strategy for significantly increasing BA.4 and BA.5 neutralizing antibodies using a bivalent vaccine fail? The most likely explanation is imprinting. The immune systems of people immunized with the bivalent vaccine, all of whom had previously been vaccinated, were primed to respond to the ancestral strain of SARS-CoV-2. They therefore probably responded to epitopes shared by BA.4 and BA.5 and the ancestral strain, rather than to new epitopes on BA.4 and BA.5. This effect could possibly be moderated by immunizing people either with BA.4 and BA.5 mRNA alone or with a greater quantity of BA.4 and BA.5 mRNA. Evidence in support of these strategies can be found in Pfizer–BioNTech’s data regarding its BA.1-containing bivalent vaccine, which showed that BA.1-specific neutralizing-antibody responses were greater in persons who were injected with a monovalent vaccine containing 30 μg or 60 μg of BA.1 mRNA or a bivalent vaccine containing 30 μg of BA.1 mRNA and 30 μg of ancestral-strain mRNA than in those who received a bivalent vaccine containing 15 μg of each type of mRNA.”
Second, vaccine fatigue is considerable, and messages patients are receiving are confusing. This contributes to the lack of uptake. Because the benefit of these vaccines is most related to time from the original COVID-19 vaccine, more frequent doses are being recommended as science changes rapidly. In addition, patients are trying to “time the virus,” so to speak, waiting to receive booster doses just before times of perceived highest transmission, such as peak winter weather.
Third, significant immunity has accrued over the past 3 years from natural infection. For example, in mid-2021, published data from the United Kingdom demonstrated an approximately 90% seroconversion rate in adults. Studies have not established how long immunity from infection lasts, but it likely wanes over time, similar to vaccination. In addition, a number of patients have received primary vaccination with an mRNA vaccine with or without the ancestral booster and have been infected with COVID-19 one or more times. Therefore, many patients have opted out of receiving a bivalent vaccine, even when they are presenting for other vaccinations. Organizational and governmental messaging has not convinced patients that vaccination prevents severe disease (hospitalization and death).
To improve messaging to patients, the CDC has begun a “Let’s RISE” campaign to encourage patients (all ages) to update their current routine vaccination status, including COVID-19 vaccines. Rise stands for Routine Immunizations on Schedule for Everyone. With the pandemic restrictions, many missed appointments, including routine annual visits, placed many children and adults at risk for significant preventable diseases including COVID-19. The Let’s RISE website has several useful resources for promoting good preventive health through vaccination.
| Table 1. Pediatric COVID-19 Vaccine Recommendations By Age Group and Vaccination Status |
|
Bivalent Vaccine/ Age Group
|
Vaccine Status
|
Recommendation
|
|
Pfizer-BioNTech Bivalent
Children 6 months through 4 years of age
|
Has not yet begun primary vaccine series
|
Complete 2 doses of the original monovalent Pfizer-BioNTech vaccine, then receive the Pfizer bivalent vaccine ≥2 months following last dose as a substitute for the final dose of primary series
|
|
Completed first 2 doses of primary series, but has not received the third dose of the primary series
|
Receive Pfizer-BioNTech bivalent vaccine ≥2 months following second dose as a substitute for the final dose of primary series
|
|
Completed original 3-dose Pfizer-BioNTech monovalent vaccine series
|
Not eligible to receive the bivalent vaccine at this time (FDA anticipates receipt of data supporting a dose of bivalent vaccine)
|
| |
Immunocompromised: completed 2 doses of monovalent COVID-19 vaccine
|
Receive dose 3 of the primary series dose using bivalent vaccine at least 2 months (8 weeks) after dose 2
|
|
Moderna Bivalent
Children 6 months through 5 years of age
|
Has not yet begun primary vaccine series
|
Complete 2 doses of the original monovalent Moderna vaccine, then receive the Moderna bivalent vaccine ≥2 months following second primary dose
|
|
Completed original 2-dose Moderna monovalent vaccine series regardless of booster status
|
Receive 1 dose of Moderna bivalent vaccine ≥2 months after previous dose
|
|
Immunocompromised: completed ≥3 doses of monovalent COVID-19 vaccine
|
Receive 1 dose of Moderna bivalent vaccine ≥2 months after previous dose
|
| Source: U.S. Centers for Disease Control and Prevention. Information should be used in conjunction with the Interim COVID-19 Immunization Schedule for Persons 6 Months of Age and Older. |
Of Concern: Bivalent Vaccine-Associated Strokes in Older Adults
The FDA and CDC use multiple safety monitoring systems to detect early safety signals for vaccines. This allows for a low threshold to facilitate further investigation as warranted. Many initial signals end up being due to factors other than the vaccine investigated.
The CDC’s Vaccine Safety Datalink (VSD), a real-time surveillance system, recently met the threshold for further evaluation of the Pfizer/BioNTech bivalent COVID-19 vaccine. This evaluation was specifically for ischemic stroke in patients 65 years of age or older and currently is not applicable to the Moderna bivalent vaccine. The timeframe for inquiry is the first 21 days following vaccination compared to days 22-42 following bivalent booster vaccination. A number of other analyses have not demonstrated this signal, including a preliminary Veterans Health Administration database evaluation as well as a Centers for Medicare and Medicaid Services database. Countries outside of the United States using the Pfizer/BioNTech bivalent vaccine have not observed this risk to date.
While the totality of the data suggests that this signal is unlikely causal, further investigation will continue, and the current data will be discussed at the next FDA VRBPAC Meeting. Therefore, no change was recommended for vaccination in this age group when this program was prepared. Pharmacists will likely be asked about these data and should be prepared to answer questions from patients who are already in low numbers receiving the bivalent vaccine.
Intranasal Vaccine Closer to Availability?
A number of companies have been researching and evaluating alternative vaccine dosage forms, most notably an intranasal COVID-19 vaccine formulation similar to the currently available product for influenza vaccination, FluMist Quadrivalent. Proposed advantages for an intranasal vaccine are provision of enhanced immunity and cellular response through direct application to the nasal mucosa, fewer systemic adverse effects, and greater acceptability to patients with needle phobia. Current intramuscular COVID-19 vaccines induce limited mucosal immunity but widespread systemic immunity, which may explain why current vaccines protect against severe disease but are not as efficacious in preventing initial transmission.
A recent study published as a preprint by The Lancet shows promise for an intranasal SARS-CoV-2 vaccine, BBV154 (iNCOVACC). BBV154 was administered intramuscularly to nearly 3,000 healthy Indian adult volunteers residing in a randomized, open-label, phase 3 clinical trial. Participants received either 2 doses of BBV154 28 days apart or Covaxin, a licensed adenovirus vector COVID-19 vaccine, given intramuscularly 28 days apart. The primary superiority outcome was immunogenicity calculated as geometric mean neutralization antibody titers (GMT) against SARS-CoV-2 measured 14 days after the second dose. Secondary outcomes included solicited adverse effects, safety, secretory/serum immunoglobulin A (IgA) responses, and cell-mediated immune responses measured at day 42 (14 days after the second dose). BBV154 met predefined superiority criteria over Covaxin through a GMT ratio of 1.45 (95% confidence interval [CI], 1.11–1.88). It also demonstrated a higher serum neutralizing GMT against omicron BA.5 compared with Covaxin. Serum IgA titers were higher with the intranasal dosage form with equivalent T cell responses between vaccines. Tolerability was excellent with both dosage forms, with approximately 5% of participants receiving BBV154 experiencing nasal side effects but only 2.7% experiencing systemic side effects.3
iNCOVACC was approved in India in December 2022 both as a primary vaccine as well as a booster. The intranasal dosage form is administered as the 2-dose series with 4 drops into each nostril for a total volume of 0.5 mL on days 1 and 28.
China also has alternative dosage forms to intramuscular injection, including an inhaled vaccine.
Whether an intranasal vaccine will be authorized in the United States in the near future is unknown. As new dosage forms are developed, they should be studied for immunogenicity but also for important clinical outcomes such as transmissibility and severe disease prevention. Transmissibility prevention would be a welcome finding in this era of vaccine hesitancy and lack of impact on transmission by currently available vaccines. In addition, the currently authorized and/or approved vaccines outside the United States are adenovirus modalities, which more closely resemble “traditional” vaccines that have garnered greater acceptance by patients, including those hesitant to receive vaccines made with newer platforms.
Use of Tixagevimab/Cilgavimab No Longer Authorized
As XBB.1.5 and other nonsusceptible subvariants became dominant among circulating strains in the United States, FDA acted to restrict the use of tixagevimab co-packaged with cilgavimab (Evusheld, AstraZeneca). As a result, this product is not authorized in the United States until further notice.
Tixagevimab/cilgavimab initially became available under an FDA emergency use authorization (EUA) for preexposure prophylaxis in high-risk populations. Patients at high risk include those with moderate-to-severe immunocompromise and those unable to receive COVID-19 vaccination because of a history of severe adverse reactions to an active or inactive ingredient of a COVID-19 vaccine product.
FDA revised this authorization on January 26, 2023, by limiting use of tixagevimab/cilgavimab to time periods when the combined frequency of nonsusceptible SARS-CoV-2 variants nationally is less than or equal to 90%. That criterion is not currently met in the United States, and hence, tixagevimab/cilgavimab is not currently authorized for use. Facilities and providers should retain unexpired product since it could be used if or when susceptible strains are again dominant.
TREATMENTS FOR COVID-19 IN THE AMBULATORY SETTING
Effectiveness of Ritonavir-Boosted Nirmatrelvir vs. VV116
Ritonavir-boosted nirmatrelvir (Paxlovid, Pfizer) has shown promising results since its EUA in December 2021. This protease inhibitor combination can be taken by ambulatory patients to prevent progression to severe disease. It is currently recommended in the NIH COVID-19 Treatment Guidelines as a first-line therapy for nonhospitalized adults and pediatric patients aged 12 years or older who have mild-to-moderate COVID-19 and a high risk of disease progression and are within 5 days of symptom onset. Ritonavir-boosted nirmatrelvir can be used in patients hospitalized with a diagnosis other than COVID-19 when they meet these 3 criteria.4
The use of ritonavir-boosted nirmatrelvir is under scrutiny for a number of reasons. First, most patients have received at a minimum a primary series of mRNA COVID-19 vaccine. A number of these primary vaccinated patients have also been infected with COVID-19 and thus have increased immunity in addition to that provided by vaccination. Most data demonstrating effectiveness of ritonavir-boosted nirmatrelvir came from primarily unvaccinated populations. In addition, the ritonavir component of the product is involved in numerous clinically significant drug interactions, making the risk/benefit assessment more complicated. Some of these interactions involve narrow therapeutic index medications, such as anticoagulants (e.g., apixaban) and immunosuppressants (e.g., tacrolimus) that may require dose adjustment or cannot be stopped during the 5-day regimen of ritonavir-boosted nirmatrelvir.
These problems leave room in the COVID-19 armamentarium for an oral agent with evidence of good outcomes in a primarily vaccinated populations and is involved in few problematic drug interactions. A recent study published in the New England Journal of Medicine evaluated a new compound named VV116 compared to ritonavir-boosted nirmatrelvir in the management of patients with mild-to-moderate COVID-19 who are at high risk of progression to severe disease. VV116 is a deuterated (a hydrogen at the 7-position of the purine ring is replaced with deuterium) remdesivir hydrobromide compound that allows for oral absorption of the drug. Similar to IV remdesivir, few drug interactions are expected with VV-116. Participants in each arm of the noninferiority trial received a 5-day course with VV116 600 mg by mouth every 12 hours on day 1 followed by 300 mg orally every 12 hours on days 2 to 5 or ritonavir-boosted nirmatrelvir 300 mg/100 mg orally every 12 hours for 5 days. Based on a primary endpoint of time to sustained clinical recovery through day 28, noninferiority was established for VV116 with a hazard ratio of 1.17 (95% CI 1.01–1.35). The median time to symptom resolution was 4 days with VV116 and 5 days with ritonavir-boosted nirmatrelvir. No participants progressed to severe COVID-19 or died from COVID-19 during the study.5
One interesting aspect of this trial was that about one-fourth of participants on ritonavir-boosted nirmatrelvir reported dysgeusia, a metallic, bitter, or other unwanted taste that is likely related to the ritonavir. This adverse effect, commonly called “Paxlovid mouth,” was covered extensively in the last COVID-19 update. Adherence remained high despite the dysgeusia, perhaps because of the emphasis on taking doses as instructed in this clinical study. In practice, prudent counseling of patients regarding this side effect is crucial to maintain adherence.5
This trial was conducted in Shanghai, China, during an outbreak of COVID-19 in March through June 2022. Nearly 75% of participants had received at least a primary vaccination series. Nearly half of patients had received a boosted vaccine course. However, the products were not mRNA vaccines as are used more commonly in the United States and many other countries. Since COVID-19 vaccines available in China have demonstrated less effectiveness than mRNA vaccines, it is surprising that no deaths occurred among the 771 trial participants. The lack of herd immunity in China further complicates interpretation of trial results, as does the fact that participants were hospitalized even when their symptoms were mild-to-moderate. Such individuals elsewhere would likely have been ambulatory.
Nevertheless, this trial demonstrates that VV116 appears to be a safe and effective option for people with mild-to-moderate COVID-19, and it has the advantage of minimal drug interactions relative to ritonavir-boosted nirmatrelvir. Longer and larger clinical trials are necessary before the place of VV116 in COVID-19 therapeutics becomes clear.
SARS-CoV-2 Testing No Longer Required
In a major adjustment in the use criteria for ritonavir-boosted nirmatrelvir and molnupiravir, positive SARS-CoV-2 test results are no longer required for before prescribing these oral agents for the management of mild-to-moderate COVID-19 in patients at high risk for progression to severe disease. In making this change on February 1, 2023, FDA said some people are testing negative even though they have developed COVID-19 symptoms after a recent close contact with a SARS-CoV-2–confirmed patient (e.g., household member). The FDA still recommends SARS-CoV-2 testing when possible to confirm the disease before prescribing these agents.
TREATMENTS FOR SEVERE COVID-19
IL-1 Antagonist Anakinra Receives EUA
In November 2022, the FDA issued an EUA for anakinra (Kineret, Swedish Orphan Biovitrum) that permits use in hospitalized patients with SARS-CoV-2 requiring supplemental oxygen for pneumonia at risk of progressing to respiratory failure. These respiratory symptoms can likely be attributed to increased serum concentrations of plasma soluble urokinase plasminogen activator receptor (suPAR), a biomarker for hyperinflammation strongly associated with adverse COVID outcomes.
Anakinra is a human recombinant interleukin-1 (IL-1) antagonist with full FDA approvals for treating rheumatoid arthritis, IL-1 receptor antagonist deficiency, and cryopyrin-associated periodic syndromes. IL-1 is a proinflammatory cytokine that causes chronic inflammation when it is overproduced or uninhibited in patients with certain conditions. Increases in IL-1 have also been detected in COVID-19 patients leading to tissue injury, especially when additional inflammatory cytokines are recruited to the lungs and further damage tissues.
Anakinra is a subcutaneous injection delivered in prefilled 100-mg syringes. The dosing for anakinra in patients with severe COVID-19 is 100 mg given subcutaneously once daily for 10 days. Because Anakinra is primarily renally excreted, clinicians should consider decreasing the dose in patients with severe renal insufficiency or end-stage renal disease (creatinine clearance <30 mL/min, as estimated from serum creatinine levels). The EUA suggests 100 mg every other day over the 10-day period, for a total of 5 doses, in these patients.
Adverse effects of anakinra include injection site reactions, elevated liver enzymes, neutropenia, and rash (incidence >1%). Anaphylactic reactions are possible when patients are sensitive to the drug, proteins derived from Escherichia coli, or components of the drug product. Immunosuppression induced by anakinra can lead to increased incidence of infection. The drug’s adverse effect profile has been well documented in use for FDA-approved indications mentioned above.
While other studies explored the use of IL-1 antagonists in treatment of COVID-19, the phase 3 SAVE-MORE trial was ultimately used to support overall effectiveness and safety when FDA issued this EUA. Patients (n = 594) included in this study had moderate-to-severe COVID-19–associated pneumonia with suspected suPAR levels ≥6 ng/mL based on clinical risk factors such as organ dysfunction and increased neutrophil counts.6
Participants who received anakinra had a statistically significantly 63% decrease in risk of progression to severe disease (odds ratio, 0.37 [95% CI), 0.26–0.50]). Mortality was also lower in patients receiving anakinra relative to standard care (hazard ratio, 0.48 [95% CI, 0.22–1.04]) at day 28 as well as day 60 (hazard ratio, 0.56 [95% CI, 0.30–1.04]. These mortality findings were not statistically significant potentially due to lack of power from low overall death rates. An important note when interpreting these results is that most patients (~85%) were receiving systemic dexamethasone therapy which has also demonstrated decreased mortality when treating COVID-19.6
Critically ill patients requiring mechanical ventilation were excluded from the SAVE-MORE trial, and this limits application of the data to this important population. The investigators also discussed the challenges of applying this study results to healthcare systems nationwide due to individual protocols of hospital systems. A key element of the study was use of suPAR levels for early identification of patients with COVID-19 pneumonia at risk of progression.6
Since suPAR assays are not commercially available in the United States, an alternative patient identification method was needed. As listed in the EUA fact sheet for anakinra, criteria for suspected suPAR levels of 6 ng/mL or more include presence of 3 or more of the following 8 criteria:
-
Age ≥75 years
-
Severe pneumonia by World Health Organization criteria
-
Current or previous cigarette use
-
Sequential organ failure assessment, or SOFA, score ≥3
-
Neutrophil-to-lymphocyte ratio ≥7
-
Hemoglobin ≤10.5 g/dL
-
History of ischemic stroke
-
Blood urea nitrogen ≥50 mg/dL and/or medical history of renal disease
Data for use of anakinra in pediatric and pregnant patients are limited. The consensus at this time is to avoid use in pregnant patients and to consult with a multidisciplinary team when considering use in children.
Interleukin-6 Antagonist Approved by FDA for Treatment of COVID-19
Monoclonal antibodies have played an important role in the management of COVID-19 over the course of the pandemic, including prevention (ritonavir-boosted nirmatrelvir), treatment of mild-to-moderate disease (e.g., sotrovimab), and treatment of severe disease (tocilizumab). Currently, the role of monoclonal antibodies has been restricted to the treatment of hospitalized patients with COVID-19.
In December 2022, tocilizumab (Actemra, Genentech) became the first monoclonal antibody approved by FDA for the treatment of severe COVID-19. It had been previously authorized by the FDA for this use. Tocilizumab is indicated for the treatment of severe COVID-19 in adult patients who are receiving systemic corticosteroid therapy (e.g., dexamethasone) and are requiring supplemental oxygen therapy, noninvasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO). The ultimate conversion from EUA to FDA approval was based on 4 primary studies. Efficacy was demonstrated with tocilizumab primarily in the EMPACTA and RECOVERY trials.7,8 The COVACTA and REMDACTA studies did not demonstrate benefit for tocilizumab but were used for further safety data evaluation.9,10
Tocilizumab remains authorized for use in patients 2–18 years of age but has not yet been approved by FDA for this age group. The EUA specifies that children and adolescents in this age range must be receiving systemic corticosteroids and requiring supplemental oxygen, noninvasive or invasive mechanical ventilation, or ECMO.
LONG COVID UPDATE
COVID-19 mRNA vaccines significantly decrease the risk of hospitalization and death from severe disease. In addition, many safe and effective intravenous and oral therapies for both hospitalized and ambulatory patients have significantly improved outcomes, including prevention of admission to the hospital. However, an area of significant concern going forward is the long-term effects of COVID-19 that linger for months after the acute phase of infection has abated. Long COVID sequalae have been reported in several organ systems, including central nervous system (e.g., brain fog) symptoms and nonspecific shortness of breath and/or fatigue. Because of these concerns, studies of longer duration are being completed to provide more details on long COVID.
A recent publication provides a comprehensive review on long COVID, detailing its effect as a multisystemic illness.11 Several postulated mechanisms contribute to long COVID, including immune dysregulation, microbiota dysbiosis, autoimmunity and immune priming, endothelial and blood clotting pathology, and abnormal neurological signaling. These mechanisms could be manifested through dysautonomia (failure of the autonomic nervous system) as well as multisystem organ involvement. Dysautonomia has been associated with other postviral illnesses, so is not an unsurprising finding in people who have had COVID-19. In addition, long COVID is also associated with myalgic encephalomyelitis (ME) and chronic fatigue syndrome (CFS) as well as postural orthostatic tachycardia syndrome. Most disconcerting regarding ME and CFS is the potential for these diseases to persist for months, years, or even lifelong. There are no safe and effective treatments for COVID-19–related ME or CFS at this time.11
Long COVID symptoms can also manifest in children/adolescents, with symptoms such as headache, dysgeusia, mental exhaustion, and sleep issues. These are 2 to 36 times more likely in adolescents aged 15–19 years of age compared with controls. High-quality research studies have proven difficult in this age group due to testing conundrums. Children infected with SARS-CoV-2 are less likely to test positive via polymerase chain reaction despite showing demonstrable seroconversion several weeks later. Estimates are that up to 90% of cases in children are initially missed. Children also are more likely to have a waning antibody response months after initial infection compared with adults.12 More research is needed to quantify these risks.
CONCLUSION
As estimated by Grabenstein, pharmacists and pharmacy technicians performed more than 42 million COVID-19 tests, provided more than 270 million vaccinations (including 8.1 million for long-term care residents), and administered more than 50 million influenza and other vaccinations, all of which minimized the burden on the health care system. These interventions plausibly averted more than 1 million deaths, more than 8 million hospitalizations, and $450 billion in health care costs.13
As the public health emergency ends for the COVID-19 pandemic, the importance of the expanded role for pharmacists must be remembered. Many states have incorporated this expanded scope of practice into their laws and regulations, and the profession should use these remarkable accomplishments to trumpet its value in other states and at the national level
REFERENCES
-
Wang Q, Bowen A, Valdez R, et al. Antibody response to omicron BA.4–BA.5 bivalent booster. Letter. N Engl J Med. 2023;388:567–569. doi: 10.1056/NEJMc2213907
-
Offit PA. Bivalent Covid-19 vaccines — a cautionary tale. Perspective. N Engl J Med. 2023;388:481–483. doi: 10.1056/NEJMp2215780
-
Singh C, Verma S, Reddy P, et al. Immunogenicity and tolerability of BBV154 (iNCOVACC®), an intranasal SARS-CoV-2 vaccine, compared with intramuscular Covaxin® in healthy adults: a randomised, open-label, phase 3 clinical trial. Available at SSRN: https://ssrn.com/abstract=4342771 or http://dx.doi.org/10.2139/ssrn.4342771
-
COVID-19 Treatment Guidelines Panel. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. National Institutes of Health. Available at https://www.covid19treatmentguidelines.nih.gov/. Accessed February 16, 2023.
-
Cao Z, Gao W, Bao H, et al. VV116 versus nirmatrelvir-ritonavir for oral treatment of Covid-19. N Engl J Med. Published online December 28, 2022. doi: 10.1056/NEJMoa2208822. Accessed February 16, 2023.
-
Kyriazopoulou E, Poulakou G, Milionis H, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med. 2021;27:1752-1760. https://doi.org/10.1038/s41591-021-01499-z
-
Salama C, Han J, Yau L, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. 2021;384:20–30. doi: 10.1056/MEJMa2030340
-
RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397(10285):1637–1645.
-
Rosas IO, Bräu N, Waters M, et al. Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N Engl J Med. 2021;384:1503–1516. doi: 10.1056/NEJMa2028700
-
Rosas IO, Diaz G, Gottlieb RL, et al. Tocilizumab and remdesivir in hospitalized patients with severe COVID-19 pneumonia: a randomized clinical trial. Intens Care Med. 2021;47:1258–1270. https://doi.org/10.1007/s00134-021-06507-x
-
Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023 Jan 13. doi: 10.1038/s41579-022-00846-2.
-
Toh ZQ, Anderson J, Mazarakis N, et al. Comparison of seroconversion in children and adults with mild COVID-19. JAMA Netw Open. 2022;5:e221313. doi:10.1001/jamanetworkopen.2022.1313
-
Grabenstein JD. Essential services: quantifying the contributions of America’s pharmacists in COVID-19 clinical interventions. J Am Pharm Assoc. 2022;62:1929–1945.e1. doi: 10.1016/j.japh.2022.08.010