ADVERTISEMENT

The Role of the Pharmacist in Managing Patients With X-Linked Hypophosphatemia
INTRODUCTION
Disorders of calcium and phosphorus are encountered in a wide variety of clinical situations across the
lifespan. One of these, hypophosphatemia, is observed in 2% to 5% of hospitalized patients and is very
common among those with alcohol use disorders.1
A rare genetic condition, X-linked hypophosphatemia (XLH), is considered the prototypic disorder of
renal phosphate wasting. In patients with XLH the body’s stores are not conserved by the kidneys in
patients with this disorder. Traditionally managed with vitamin D and supplements of calcium and
phosphate, research into the pathophysiologic causes of XLH is yielding more specific treatments that
promise to improve bone physiology and prevent sequelae in those with this condition.2
In this program, the underlying functions of phosphate, genetic causes and pathophysiology of XLH,
presentation and symptoms of XLH, and current and emerging approaches to clinical management of
XLH are reviewed.
CALCIUM, PHOSPHORUS, AND VITAMIN D PHYSIOLOGY
In the human body, calcium and phosphorus homeostasis is affected by many different hormonal,
nutritional, and physiologic factors that regulate intestinal absorption, influx and efflux from bone, and
kidney excretion and reabsorption. While a common constituent of the body and the major intracellular
anion, phosphorus is also covalently bound in organic phosphate esters in genetic material and
metabolic enzymes. A small amount of intracellular inorganic phosphate is involved in a critical function
throughout the body — regeneration of the storage molecule for energy, adenosine triphosphate (ATP).3,4
The limited amount of inorganic phosphate available in the extracellular space is also important in a
wide variety of normal body functions. Extracellular inorganic phosphate (which is measured when
serum phosphorus levels are assayed) is the primary determinant of intracellular phosphate; it is
involved in metabolic regulation, hydrogen ion shifts, and balance among vitamin D, serum calcium, and
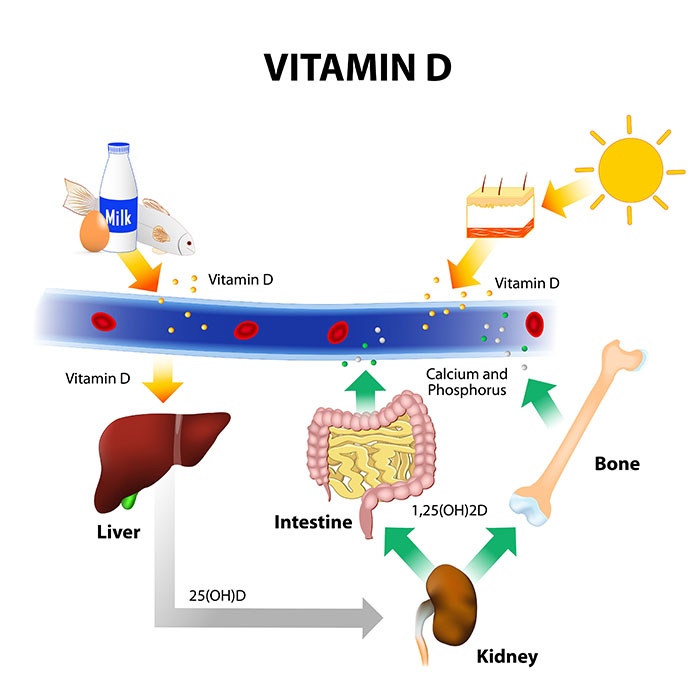
levels of the hormones calcitonin, cortisol, and parathyroid hormone. As depicted in Figure 1, serum
phosphorus levels can affect and be affected by bone, kidney, and intestine activity.2,4
Figure 1. Interplay among intracellular and extracellular phosphorus stores of the body.
 |
Phosphorus is consumed by most people as part of the normal diet of dairy products, meat, and
vegetables. Serum phosphorus concentrations can vary by 2 mg/dL throughout the day based on
carbohydrate intake, insulin secretion, and diurnal variation. The traditional model of gastrointestinal
absorption of phosphorus has emphasized the actions of vitamin D and parathyroid hormone (PTH) for
increases in mineral absorption, but passive absorption also occurs.3,5
Phosphorus and phosphate are excreted renally. Following glomerular filtration, 85% to 90% of the ions
are normally reabsorbed in the proximal renal tubule by passive transport coupled to the positive
sodium ion. Reabsorption is inhibited by PTH and the active form of vitamin D3, 1,25-dihydroxyvitamin
D3 (calcitriol); growth hormone, insulin, and insulin-like growth factor 1 increase phosphate
reabsorption in the renal tubule. As shown in Figure 2, phosphate transport is regulated by fibroblast
growth factor 23 (FGF23); its action depends on a cofactor, transmembrane protein klotho.3,4,6
HYPOPHOSPHATEMIA
Normal serum phosphorus levels vary according to age. Hypophosphatemia is defined as serum
phosphorus concentrations less than 4 mg/dL in children younger than 12; the normal range gradually
decreases during adolescence, and the lower end of the normal range in adults is 2.5 mg/dL. The
condition is generally asymptomatic until levels reach about 1 mg/dL in adults. Hypophosphatemia can
be acute or chronic. When hypophosphatemia is severe, symptoms result from reduced levels of 2,3-diphosphoglycerate in erythrocytes and intracellular ATP levels in virtually all organ systems. The
neurologic system is particularly affected, with symptoms of irritability, apprehension, weakness,
numbness, paresthesia, and confusion. If not corrected, severe hypophosphatemia can lead to seizures
or coma. Given the role of phosphates in bone modeling, osteomalacia and osteopenia are other
potential outcomes of deficiency states.4
X-linked hypophosphatemia
XLH is a rare genetic disorder with an estimated incidence of 1 in every 20,000 live births. It has been
referred to as vitamin D–resistant rickets in the past. The X-linked dominant mutation occurs in the
phosphate regulating endopeptidase homolog, X-linked (PHEX) gene (Xp22.1). It most often presents in
children between 6 months and 2 years of age as lower-extremity bowing, short stature, and over time,
joint pain and impaired mobility. Because the mutation is dominant (one mutant gene is sufficient to
produce the condition), it can occur in females as well as males. Girls born to a father with the condition
are certain to have XLH, since the hemizygous father would by definition transmit a defective, dominant
gene.7
As a genetic disorder, XLH is sometimes diagnosed before phenotypic expression through specialized
testing in individuals who have family members with the disease. Children can also present initially with
dental abscesses. Since these are not caused by dental caries, the teeth may have a normal external
appearance. In adults with the disease, hearing impairment and arthritis are common as a result of bone
abnormalities.2,8,9
PATHOPHYSIOLOGY
In XLH, loss-of-function mutations on the gene that codes for PHEX results in overproduction of FGF23
by the osteocyte/osteoblast. This peptide hormone regulates circulating phosphate levels by reducing
renal tubular phosphate reabsorption and inhibiting renal vitamin D production. Elevated FGF23 results
in excess phosphate excretion from the kidney as well as decreased synthesis of 1,25-dihydroxyvitamin
D3, which reduces active phosphate absorption in the gastrointestinal tract (Figure 2).9
Conditions that may acutely exacerbate the phosphate wasting seen in patients with XLH include
prematurity, malignancy, diseases of organs associated with vitamin D and calcium metabolism such as
kidney disease or liver and biliary tract disease, and rarely malabsorption syndromes in conditions such
as celiac disease or cystic fibrosis.10 Medications such as loop diuretics, corticosteroids, anticonvulsants
(phenytoin), and antacids that contain aluminum (due to the binding of aluminum to phosphates) may
also exacerbate XLH signs and symptoms.
Without effective treatment, chronic hypophosphatemia in patients with XLH can lead to rickets and
osteomalacia, stunted growth, persistent short stature (adult height of 4 feet or less), lower-limb
deformity, pain, and physical dysfunction that can limit daily activities. Patients sometimes need
corrective surgery for severe bowing or tibial torsion unlikely to be corrected by medical management.
To prevent problems in the mouth, patients need to adhere to a rigorous oral hygiene regimen and
regular schedule of professional check-ups.2,7,9
Case study 1: Pediatrics XLH presentation
MJ is a 19-month-old, 10.7-kg female toddler, coming in for her 18-month immunizations at her
pediatrician’s office. Her mother complains that MJ has not wanted or tried to stand or walk unlike her
two older brothers, and when she does try, she falls over immediately, not able to stand for more than a
few seconds. She also notes that MJ’s legs seem to bend and arch out at the knees. MJ was born full
term and delivered vaginally, without any medical concerns otherwise. Her diet is normal for her age
with formula and solid foods. Her vaccinations are up to date, including influenza last year. All other
developmental milestones are within normal range including speech, vision, hearing, and cognition. MJ’s
weight, height, and head circumference measured at the 30th, 5th, and 30th percentiles, respectively.
Musculoskeletal examination is notable for bowing of the legs, and X-ray imaging shows the bone
structure depicted in Figure 3. Pertinent laboratory findings include serum calcium within normal limits,
serum phosphorus 2.8 mg/dL (reference range provided by the laboratory, 3.8–6.5 mg/dL), elevated
serum alkaline phosphatase (ALP), and decreased tubular reabsorption of phosphate (TRP) corrected for
glomerular filtration rate (TRP/GFR) of 2.0 mg/dL (reference range 2.9–6.5 mg/dL).
Clinical Presentation
Rickets, osteomalacia, and growth retardation occur as a result of XLH. XLH is the most common genetic
cause of rickets and osteomalacia. The most prominent clinical feature of XLH is bowing deformities of
the legs (Figure 4); however, the symptoms of XLH vary widely depending on the severity of the
condition. Some people may present with bone-related symptoms such as bone pain and joint pain,
while others may present only with hypophosphatemia.11 Because XLH is a rare disease and has a
genetic etiology, it may not be diagnosed or suspected until other causes of hypophosphatemia (e.g.,
vitamin-dependent hypophosphatemia) are ruled out.
Typical symptoms of XLH include slowed growth rate in the first year of life, beginning to walk late or
not walking beyond the reasonable age, bone pain, unstable gait, joint pain caused by calcification of
tendons and ligaments (enthesopathy), stress fractures, muscle pain and weakness, dental abscesses
and pain, abnormal dental development, and rickets despite supplemental treatment with vitamin D.2,11
Some patients may also present with pain flaring of wrists or ribs at the diaphragm, fractures, skull
deformity, osteopenia, or scoliosis.10
Children with XLH typically present with bowed legs, medial tibial torsion (widened legs arching out on
the knees), and short stature. The structural symptoms are noticeable within the first 1 to 2 years of life
when a child starts to walk and bowing of the weight-bearing long bones become apparent. Due to this
early-stage presentation of bone-related symptoms, XLH is often misdiagnosed as rickets of vitamin D
deficiency.
Dental and periodontal defects are common in patients with XLH. Because of improper development of
dentin, enamel, and pulp related to low phosphate levels, dental abscesses can occur due to
spontaneous infection of the dental pulp tissue, without any trauma or decay. However, patients’ teeth
look normal clinically, making the diagnosis and treatment of the abscesses complicated.12
Craniosynostosis may be present, manifested by signs of elevated intracranial pressure such as
headaches, vomiting, papilledema, or bulging of the anterior fontanel.13 In adult patients, case reports of
enthesopathy of vertebral ligaments, spinal cord compression and paraplegia, and spinal stenosis reflect
the significant joint deformity and impaired mobility that can accompany this condition.11,14
Sensorineural hearing may be affected for some patients, but this typically presents during adulthood in
treated patients.11,14 Patients may have mild-to-severe sensorineural hearing loss, which typically affects
low and high frequency sounds. Some patients may complain of tinnitus and vertigo associated with low
frequency hearing loss.12
Once a patient’s family history is taken (it is important to ask about parents or siblings with short stature
and rickets), imaging of the knees needs to be performed to assess the initial bone structure and to rule
out other causes for short stature such as skeletal dysplasias.7
To assess the possibility of a diagnosis of XLH, laboratory tests are needed, including serum calcium,
phosphorus, PTH, and vitamin D assays. To confirm the diagnosis by demonstrating renal wasting of
phosphate, a 2-hour fasting urine specimen with a midpoint blood sample is used to calculate the
percentage of TRP and to determine the tubular threshold maximum for phosphate.2
Case study 2: Adult XLH presentation
BR is a 39-year-old woman who was diagnosed with vitamin D-resistant rickets after multiple attempts
to correct the symptoms with vitamin D and phosphate supplements when she was younger. She is
complaining of weakness of the lower extremities, pain in her knees and back, and hearing loss in the
left ear. She is of short stature (3 feet 10 inches) and her gait is unstable mainly due to pain on walking.
Her diet is within normal range and she denies any alcohol, tobacco, or illicit drug use. Her family history
is significant for rickets as her father and her sister both were diagnosed with vitamin D-resistant rickets,
at the ages of 5 and 2, respectively. She has not been genetically tested for XLH. She has been taking
vitamin D and phosphate supplements but admits to not taking them regularly as they do not seem to
help her pain.
Laboratory findings are within normal limits for serum calcium, serum phosphorus, PTH, growth
hormone, and insulin-like growth factor-1 (IGF-1) concentrations. 25-OH vitamin D3 and 1,25-(OH)2-vitamin D3 levels are slightly below normal. Serum ALP is slightly elevated. TRP/GFR is 1.6 mg/dL
(reference range 2.2–3.6 mg/dL).
TREATMENT
Therapeutic goals in patients with XLH vary by age at diagnosis, whether the epiphyseal plate has closed
and growth is therefore complete, and the overall severity of the condition. Some patients with XLH are relatively unaffected and do not require treatment. For most of those diagnosed as children, effective
therapies are critical in attaining a relatively normal height in adulthood and avoiding complications of
the disease. In adults with XLH, treatment is directed at reducing pain symptoms and the extent of
osteomalacia.2
The initial treatment for XLH focuses on maintaining adequate levels of phosphate and vitamin D. To
achieve this goal, phosphate supplements and high-dose vitamin D have been the mainstay of therapy
for XLH. Calcitriol increases calcium and phosphorus absorption and increases mineralization of the
bone indirectly via increased calcium absorption in intestinal lumen.10 Treatment is typically initiated
upon diagnosis, usually in early childhood, as this has been associated with better outcomes for XLH.
Treatment should be continued through adolescence (when growth has stopped) in mild cases or
through adulthood in most cases. However, treatment regimens with phosphate supplements are
complicated by the need for multiple daily dosing and the use of various formulations. Additionally,
these treatments do not address the underlying cause of reduced phosphate reabsorption, and their use
is associated with treatment-limiting adverse effects such as hypercalcemia, elevated PTH, and
nephrocalcinosis.7
Maintenance treatment has been shown to slow progression of bowing, promote growth, progressively
correct leg deformities, facilitate tooth mineralization, and reduce the need for corrective surgery.7,12 It
is important to treat patients with both phosphate and calcitriol because treatment with phosphate
alone increases the risk for hyperparathyroidism. However, normalization of the serum phosphorus
concentration is not a therapeutic goal for XLH because it frequently indicates overtreatment and
increases the risk for treatment-related complications.11
A new monoclonal antibody, burosumab, targets elevated levels FGF23 and was recently approved for
treatment of XLH of adult and pediatric patients 1 year of age or older. Clinical trials have demonstrated
efficacy and safety in adults and children without complications of phosphate therapy.9
Children
Medical therapy
The goal of treatment is to decrease bone pain (response expected within a few weeks), normalize the
serum ALP level in 6–12 months, increase in growth velocity in about 1 year, and straighten the legs in
3–4 years (1-cm decrease in intercondylian distance [between the medial and lateral condyle at the
knee] or in intermalleolar distance [between the two malleoli at the two ankles], checked every 6
months).12 The treatment focuses on maintaining adequate levels of phosphate and vitamin D through
supplementation with oral phosphate administered 3 to 5 times daily and high-dose calcitriol.
Oral phosphate salts: The dose for phosphate supplementation varies depending on the severity of the
disease and developmental stage of the patient. In infants, the dose is 55–70 mg/kg per day divided in 3
to 5 doses. The starting dose for infants and children is typically 40 mg of elemental phosphorus/kg/day;
in addition to dosing during the child’s waking hours, a nighttime dose is recommended to achieve
satisfactory results.16 If there is no improvement in the growth within the first year of therapy, the dose
can be titrated upward based on age, weight, and clinical condition. For adolescents in puberty, the dose
is 35–50 mg/kg per day divided in 3 doses per day.2,12
Vitamin D: Calcitriol is the synthetic analog of the active form of vitamin D, 1,25-dihydroxyvitamin D3.
The dose is 20–30 ng/kg/day administered in 2 to 3 divided doses to 50–70 ng/kg/day up to a maximum dose of 3 mcg daily with the phosphate.11 While effective in increasing the absorption of calcium and
phosphorus, the use of calcitriol does not correct renal phosphate wasting in patients with XLH.15
Growth hormone (GH): GH has been used as adjunctive therapy for XLH in children in an effort to
increase the growth rate. However, reports of the long-term outcome of using GH have not been
consistent and include cases of increased serum ALP activity and worsening of leg deformities.2 There is
no adequately controlled evidence to support its use to improve adult height in a typical population with
XLH. Other limitations of GH are its cost and side effects.
Surgery
Corrective osteotomies are not routinely performed in children under 6 years of age because medical
therapy typically manages bone deformities in this early developmental age group.2 For children whose
diagnosis is delayed or whose initial treatment is not successful, osteotomy can be considered to align
severely distorted legs. In prepubertal children who have not yet completed their full growth velocity,
surgical therapy can be considered using the least invasive option. However, it is associated with
potential for prematurely stopping growth.2,11 Newer and less invasive approaches include
epiphysiodesis, which induces corrective differential growth of the growth plate.2
Dental care
Rigorous dental hygiene is a mainstay of management, including brushing 2 to 3 times daily and having
regular dental hygienist visits. Sealant application to the enamel of the teeth may be performed if
deemed necessary. Periodic dental procedures may also be necessary for spontaneous abscesses that
may occur in children.
Other interventions
Craniosynostosis (abnormal fusion of the skull) secondary to hypophosphatemic rickets is the most
common metabolic cause of congenital craniosynostosis. It can present with increased intracranial
pressure, headache, or papilledema. When craniosynostosis is suspected, the patient should be
screened and referred to a craniofacial specialist for further evaluation.13
Adults
Medical therapy
Patients who initiated therapy at the time of a pediatric diagnosis of XLH often need to continue
treatment in adulthood. Patients sometimes exhibit a lack of adherence to therapy in adulthood
because the effects of XLH are not as prominent in adulthood as they were in developmental stages. As
such, adult patients may not realize the therapeutic benefits of treatment.12 However, the
pathophysiology of phosphate wasting persists chronically in their lifetime, and patients can still benefit
from treatment. In contrast, asymptomatic patients may not benefit from continued therapy, but may
experience complications from the therapy. Thus, in adulthood, treatment should be preferentially
considered for patients with symptoms.2,12,16
As in children, the first-line agents for XLH in adults are phosphate supplements and calcitriol.2 The goal
of treatment should focus on resolving symptoms such as pain, decreasing the extent of osteomalacia,
and improving compromised function and mobility.2,16 Of note, enthesopathy leading to spinal cord
compression and pain (e.g., paraspinal enthesopathy and spinal ligament calcification) does not improve
with treatment. Treatment may be stopped with resolution of the symptoms and restarted if symptoms
recur. Since symptom resolution is the therapeutic goal for adult patients, treatment should be
discontinued if symptoms do not improve within 1 year.2
As discussed further in the Newer Treatments section, adult patients have benefited from therapy with
burosumab. Its place in therapy in adults with XLH who fail phosphate/calcitriol treatment is not yet
clear.
Calcitriol: When treatment is being started or restarted, calcitriol is given about 1 week before
phosphate supplementation begins. This decreases the risk of exacerbating pre-existing secondary
hyperparathyroidism, or inducing the condition when calcitriol increases the serum concentration of
phosphate while causing a further depletion of serum calcium (see Complications of treatment).
During this initial week before phosphate therapy, calcitriol is given at a dose of 0.5–0.75 mcg/day in 2
divided doses for patients with normal calcium levels and normal or only mildly elevated serum PTH.2
Oral phosphate salts: For patients with normal calcium levels and normal or mildly elevated serum PTH,
daily elemental phosphorus 250 mg should be started after 1 week of treatment with calcitriol. The dose
should be titrated every 4 days to 750–1,000 mg of elemental phosphorus.2
Cinacalcet: In patients with hyperparathyroidism (i.e., overactive parathyroid gland in response to low
serum calcium level), particularly those with rising serum PTH and ALP levels, the calcimimetic agent
cinacalcet is used to normalize the serum PTH level. The initial dose is 30 mg at bedtime with increases
to a maximum dose of 60 mg at bedtime. However, outcomes have varied; some patients have had
refractory hyperparathyroidism.2
Pregnant women: There are currently no evidence-based recommendations on the use of phosphate
and calcitriol in pregnant women with XLH. However, if a woman with XLH is on an active maintenance
therapy at the time of conception, she should be continued on treatment through the pregnancy with
careful monitoring of urinary calcium-to-creatinine ratios to monitor hypercalciuria for any needed
changes to the therapy.11,16
Surgery
Some patients continue to experience persistent lower-limb bowing and torsions despite medical
treatment and may require surgical treatment. In older children of postpubertal age or adults, surgical
procedures may include joint replacement surgery, especially of the knee or hip. A total hip and knee
arthroplasty may be required for some patients because of degenerative joint disease and
enthesopathy. Patients should begin phosphate and calcitriol supplementation 3 to 6 months before the
surgical procedure and continue it for 6 to 9 months after.2
Dental care
Patients with XLH are susceptible to recurrent dental diseases and abscesses; these can result in
numerous root canals and tooth extractions, leading to premature loss of permanent teeth. Consistent
and thorough oral hygiene with flossing and regular dental care including fluoride treatments is
necessary to prevent dental problems. Pit and fissure sealants have been recommended without a good
amount of evidence, and no specific therapy has been demonstrated to prevent dental complications.2,12
Sensorineural hearing
Patients with complaints of hearing loss, tinnitus, or vertigo should be referred to a specialist for hearing
tests as well as for management options. These may include hearing aid, vibrotactile devices, and
cochlear implantation.11
Complications of treatment
The major complications from long-term treatment with phosphate and calcitriol supplementation
include gastrointestinal side effects and risk of metabolic and endocrine abnormalities such as hypercalcemia, hypercalciuria, nephrolithiasis, nephrocalcinosis, hyperparathyroidism, and possibly
chronic kidney disease.2 Among these, the two most important complications of XLH are
nephrocalcinosis and hyperparathyroidism.16
When phosphate is used unopposed or if the patient’s serum concentration is relatively high, it can
increase the risk for hyperparathyroidism. Conversely, calcitriol can increase the risk for hypercalcemia,
hypercalciuria (urinary calcium concentration >4 mg/kg/day or calcium/creatinine ratio >0.7 in the first
year of life or above 0.3 after 1 year), and nephrocalcinosis if used unopposed or the patient’s serum
concentration is high.2,11,17
Hyperparathyroidism may occur at any time in the course of XLH, primarily because of PTH stimulation
from high doses of phosphorus (>50 mg/kg/day). This secondary hyperparathyroidism, if it persists for a
long period of time, can damage normal parathyroid function and potentially result in tertiary
hyperparathyroidism.17 Tertiary hyperparathyroidism is not very common but is a significant health risk
for intense bone resorption, nephrocalcinosis, and renal insufficiency. Contributory factors to the
progression of tertiary hyperparathyroidism include early age of initial treatment, duration of treatment,
high doses of elemental phosphorus, and very high PTH levels (~400 pg/mL).17
Nephrocalcinosis is diagnosed based on renal ultrasonography and is present in up to 80% of patients
with XLH.16 Nephrocalcinosis can develop from an excessive calcitriol dose or from nonadherence with
oral phosphate supplementation. Thus, careful surveillance of serum and urine calcium is necessary to
minimize nephrocalcinosis, and the dose of calcitriol should be reduced when hypercalcemia or
hypercalciuria occur.17 Alternatively, administration of thiazide diuretics with or without amiloride can
arrest the progression of nephrocalcinosis.16
Monitoring and dose adjustments
For patients on phosphate and calcitriol therapy, the following monitoring is recommended2,11,17:
- Quarterly monitoring of serum concentrations of phosphate, calcium, creatinine, ALP, and intact
PTH. Doses of phosphate and calcitriol should be adjusted based on (1) evidence of therapeutic
success including reduction in serum ALP activity, changes in musculoskeletal examination,
improvement in radiographic rachitic changes, and improved growth velocity; and (2) evidence
of therapeutic complications including hyperparathyroidism, hypercalciuria, and
nephrocalcinosis. For adults whose conditions are stable during long periods of treatment,
monitoring can be done every 6 to 9 months.
- If phosphorus supplementation causes diarrhea that is not tolerable, the dose of
phosphorus should be decreased by 250 to 500 mg and then gradually titrated in steps
of 125 mg. ALP is useful in assessing skeletal healing and should decrease with
treatment. If slow growth and elevated ALP persist, the phosphorus dose might not be
adequate or an issue of nonadherence may be present.
- Normophosphatemia may indicate overtreatment in some patients and may result in
secondary hyperparathyroidism.
- Hypercalcemia or hypercalciuria may indicate a need to decrease the calcitriol dose.
- PTH levels are measured to assess hyperparathyroidism secondary to treatment. PTH
levels should decrease if the calcitriol dose is increased or phosphate dose is decreased.
- Alterations in biochemical values are surrogate endpoints and may not predict skeletal
response.
- Quarterly monitoring of urinary calcium, phosphate, and creatinine for evidence of
hyperparathyroidism, increased renal phosphate or calcium excretion, and signs of chronic
kidney disease.
- Annual X-ray imaging of the lower extremities to assess skeletal response to treatment
- In children, decreased bowing and increased bone growth should be noted.
- Semiannual or annual renal ultrasound to assess for nephrocalcinosis
- Dental follow-up twice a year at minimum
- Patient counseling to avoid medications that may exacerbate or worsen rickets (e.g., antacids)
NEWER TREATMENTS
After many years of treatment of XLH with phosphate and calcitriol, novel approaches to treating XLH
have been developed within the past several years and some are currently in clinical trials. At the time
this program was prepared (summer 2018), the Clinicaltrials.gov registry listed 27 active, completed, or
future studies on XLH in the United States and abroad, including 19 that focused on pediatric patients. A
majority of these approaches target the underlying pathophysiology with FGF23 antibodies to neutralize
the effects of the increased FGF23 in XLH. Other approaches involving the FGF23 antibodies include
inhibiting downstream FGF23 signaling or competing with FGF23 receptors.12
One of the most actively studied monoclonal FGF23 antibodies is KRN23 (burosumab), a recombinant
agent that targets FGF23. Currently, 13 clinical trials are registered in Japan, South Korea, and the
United States. Clinical trials investigating burosumab began in 2008, and the agent has been approved
for use in both pediatric and adult patients in the European Union and the United States, where it was
designated an orphan drug in 2014 and 2009, respectively. Three phase 2 and three phase 3 clinical
trials have been completed to date, and two phase 2 trials are ongoing for treatment of patients with
tumor-induced osteomalacia and epidermal nevus syndrome.
The relative place in therapy of this new drug is not yet clear. Since it is a more specific treatment for the
underlying defect, burosumab could emerge as a preferred agent as clinical experience accumulates for
its use in pediatric and adult patients with XLH. An important question to be addressed is whether early
treatment can change the clinical course of this lifelong condition.
Resources for Pharmacists: Pharmacists should be aware of and if appropriate, counsel patients and caregivers that this product has a patient assistance program to assist with insurance
coverage and financial support of patients with XLH.
Evidence to date
Burosumab-twza (Crysvita) is a fully human recombinant immunoglobulin G1 monoclonal antibody that
targets FGF23. It is approved in the United States for treatment of XLH in adults and children 1 year of
age or older. It binds to and inhibits the biological activity of FGF23, thereby restoring renal phosphate
reabsorption and increasing serum concentration of 1,25-dihydroxyvitamin D3. Burosumab should not
be used (1) in conjunction with oral phosphate and vitamin D supplementation, (2) if serum phosphorus
is within or above the normal range for age, or (3) in patients with severe renal impairment or end-stage
renal disease.18
Children
Two phase 2 clinical trials investigated the efficacy and safety of burosumab in pediatric patients.9,18 In a
randomized, open-label, dose-defining phase 2 trial, 52 prepubescent XLH patients of 5–12 years of age
received either burosumab or placebo subcutaneously every 2 or 4 weeks up to a maximum dose of 2 mg/kg for at least 64 weeks. The mean serum phosphorus level significantly increased from 2.4 ± 0.4
mg/dL to 3.3 ± 0.4 and 3.4 ± 0.45 mg/dL at week 40 and week 64, respectively. The mean TRP/GFR
increased significantly from 2.2 ± 0.49 mg/dL to 3.3 ± 0.6 and 3.4 ± 0.53 mg/dL at week 40 and week 64,
respectively. The mean serum ALP significantly decreased from 462 ± 110 U/L to 354 ± 73 U/L at week 64 (23% decrease). Radiographic evaluation revealed significant improvements in mean Rickets Severity
Score (RSS) and Radiographic Global Impression of Change (RGI-C); lower RSS indicates improvements in
rickets and a change of RGI-C score of +2.0 reflects bone healing. Of 26 patients receiving burosumab
every 2 weeks, 18 achieved an RGI-C score of +2.0 or more at week 40 and maintained the scores above 2.0 at week 64. The mean height Z score improved from –1.72 ± 1.03 at baseline to –1.54 ± 1.13 at week 64. Adverse effects included injection site reaction, headache, cough, nasopharyngitis, and pain in an
arm or leg. One patient was hospitalized for fever and myalgia, which were moderate in severity and
possibly related to burosumab, one patient had severe rash, and one had a tooth abscess.9
The second study was an open-label, 64-week trial of 13 children ages 1–4 years. All of the children
received burosumab at a dose of 0.8 mg/kg every 2 weeks titrated of up to 1.2 mg/kg based on serum
phosphorus measurements for at least 40 weeks of therapy. The mean serum phosphorus level
significantly increased from 2.5 ± 0.28 mg/dL to 3.5 ± 0.49 at week 40. The mean serum ALP
concentration decreased from 549 ± 194 U/L to 335 ± 88 U/L at week 40 (36% decrease). Radiographic
evaluation revealed significant improvements in mean RSS (from 2.9 to 1.2) and mean RGI-C (+2.3 ± 0.08). All 13 patients achieved an RGI-C score of +2.0 or more. Adverse effects occurring in more than 3
participants in this small trial were pyrexia and vomiting.18
Adults
In a dose-escalation, dose-extension, open-label study of 28 adult patients with XLH, patients were given
increasing doses of burosumab every 4 weeks for a total of 4 doses and continued to receive phosphate-titrated doses once monthly for 12 months. Most patients had relatively stable doses after the eighth
dose of burosumab; 80% of them were on either 0.6 or 1 mg/kg. The main study outcomes were serum
phosphorus level and safety. From a total of 28 patients with a mean age of 41.9 years (range, 19 to 66),
26 received four doses and 19 received all 16 doses over the study. During dose escalation, TRP/GFR,
serum phosphorus, and 1,25-dihydroxyvitamin D3 levels increased, peaking at 7 days for TRP/GFR and
serum phosphorus, and at 3–7 days for 1,25-dihydroxyvitamin D3.19
After each of four escalating doses, peak serum phosphorus levels were between 2.5 and 4.5 mg/dL in 14.8%, 37.0%, 74.1%, and 88.5% of participants, respectively. During the 12-month extension, peak
serum phosphorus was in the normal range for 57.9%–85.0% of participants, and 25% maintained
trough serum phosphorus levels within the normal range. Mean serum and urinary calcium remained
within normal limits. Transient hypercalcemia (calcium >10.5 mg/dL) occurred in 2 patients and
hypercalciuria occurred in 5 patients. The urinary phosphate level varied widely among the patients.
Adverse events included arthralgia, nasopharyngitis, back pain, extremity pain, diarrhea, sinusitis, upper
respiratory infection, dizziness, headache, injection site reaction, and restless leg syndrome.19
In a randomized, double-blind, placebo-controlled study of 134 adults with XLH, patients received
burosumab 1 mg/kg every 4 weeks for 24 weeks.18 All patients had skeletal pain associated with XLH/osteomalacia at baseline, the mean age of patients was 40 years (range 19 to 66 years), and 35% of
participants were men. Oral phosphate and calcitriol treatment was not allowed during the study.
Compared with placebo group, significantly more patients from the burosumab group achieved serum
phosphorus levels above the lower limit of normal (8% vs. 94%). The mean TRP/GFR improved
significantly in the burosumab group compared with placebo at week (from 1.68 vs. 1.60 to 2.21 vs. 1.73, respectively). Radiographic evaluation of osteomalacia and active fracture/pseudofracture sites
revealed that burosumab group demonstrated a higher rate of complete healing vs. placebo group at week 24. Additionally, a total of 6 new fractures or pseudofractures appeared in 68 patients with
burosumab compared with 8 in 66 patients in the placebo group. While burosumab therapy was
associated with a significant improvement in stiffness compared with placebo at 24 weeks, there was no
significant difference in pain between the two groups.20
A 48-week, open-label, single-arm study in 14 patients with XLH assessed the effects of burosumab on
improvement of osteomalacia. Patients received 1 mg/kg injection of burosumab every 4 weeks and
other treatments were not permitted during the study. At baseline, the mean age of patients was 40
years (range 25 to 52 years) and 43% were men. After 48 weeks of treatment, osteomalacia was
improved in 10 patients, with a decrease of 57% in osteoid volume/bone volume. Osteroid thickness
declined in 11 patients from a mean of 17 ± 4.1 micrometers to 12 ± 3.1 micrometers (–33% change).
Mineralization lag time also declined in 6 patients from a mean of 594 ± 675 days to 156 ± 77 days (–
74%).18
Dosage and adverse events
For children, the starting dose of burosumab is 0.8 mg/kg rounded to the nearest 10 mg, administered
subcutaneously every 2 weeks. The minimum starting dose is 10 mg up to a maximum dose of 90 mg.
Doses may be titrated based on periodic monitoring up to about 2 mg/kg or the maximum dose to
achieve normal serum phosphorus level. For adults, the dose is 1 mg/kg rounded to the nearest 10 mg,
administered subcutaneously every 4 weeks.18
About 25% of pediatric patients experienced adverse effects of headache, injection site reaction,
vomiting, pyrexia, pain in extremity, or decreased vitamin D levels. About 5% of adult patients reported
back pain, headache, tooth infection, restless leg syndrome, decreased vitamin D level, dizziness,
constipation, and increased phosphorus levels.18
Case studies 1 and 2 (continued)
Case 1. MJ can begin therapy with oral phosphate salts and calcitriol to increase the serum phosphorus
level and decrease ALP and bone damage. With a weight of 10.7 kg, she can be prescribed 40 mg of
elemental phosphorus per kg per day in 4 divided doses and given with calcitriol 20 ng/kg twice daily. On
this regimen, monitoring is needed as follows:
- Serum concentrations of phosphorus, calcium, creatinine, ALP, and intact PTH should be
measured after 4–6 weeks of therapy to assess and adjust the doses if necessary. Hypercalcemia
or hypercalciuria may be a sign to decrease the calcitriol dose. PTH levels are measured to
assess hyperparathyroidism secondary to treatment and should decrease if the calcitriol dose is
increased or phosphate dose is decreased.
- Once stabilized, therapy can be monitored quarterly monitoring based on urinary calcium,
phosphate, and creatinine for evidence of hyperparathyroidism and increased renal phosphate
or calcium excretion.
- Annual X-ray imaging of the lower extremity to assess skeletal response to treatment decreasing
bowing should be noted.
- Monitor growth and height velocity.
- Semiannual or annual renal ultrasound should be conducted to check for nephrocalcinosis.
- Dental examinations are recommended twice a year at a minimum.
If no improvement is seen with the replacement therapy or if it is not tolerated, MJ can be considered
for treatment with burosumab, at an initial dose of 10 mg administered subcutaneously every 2 weeks (0.8 mg/kg, rounded to the nearest 10 mg). Supplemental vitamin D and phosphorus products should be
discontinued during burosumab therapy. The burosumab dose can be increased up to about 2 mg/kg to
the maximum dose to achieve an adequate serum phosphorus level based on monitoring every 4 to 6
weeks during titration. Once stable, MJ should be monitored quarterly for serum levels of phosphate,
calcium, creatinine, ALP, and intact PTH as well as radiographic evaluation to assess response to the
treatment.
Case 2. Given BR’s family history and unsuccessful vitamin D supplementation therapy, XLH is suspected.
While testing may be necessary for infants and children to begin early treatment for optimal outcomes,
it may not be necessary for adult patients who are stable with corrective therapy. However,
confirmatory tests, if indicated for XLH-specific treatment, can be done via molecular genetic testing to
determine if the PHEX variant is present.
BR should stay on the supplemental therapy with oral phosphorus and calcitriol to maintain
homeostasis, unless otherwise determined based on laboratory findings. Since she is normocalcemic
and her PTH is within normal limits, she can continue on calcitriol 0.5–0.75 mcg/day administered in two
divided doses and 250 mg/day of elemental phosphorus after 1 week of calcitriol treatment. Serum
concentrations of phosphate, calcium, creatinine, ALP, and intact PTH should be measured quarterly and
the dose adjusted as needed. Other monitoring includes semiannual or annual renal ultrasound
examination to check for nephrocalcinosis.
If no improvement is seen with the replacement therapy or if it is not tolerated, BR can be considered
for treatment with burosumab, at an initial dose of 1 mg/kg rounded to the nearest 10 mg, administered
subcutaneously every 4 weeks. Supplemental vitamin D and phosphorus products should be
discontinued during burosumab therapy.
She may also need to be referred to an orthopedic specialist for further consultation to determine if her
joint and bone conditions are worsening and assess causes of her back pain as well as to an
otolaryngologist for her hearing problem.
CONCLUSION
Medication regimens used to manage XLH are complex but can play an important role in the lives of
patients. Those on standard therapies need assistance with organizing multiple daily doses and
maintaining a positive attitude about the disease and its characteristic aches and pains. Newer therapies
are injectable agents given every 2 or 4 weeks, presenting a new set of challenges to patients and
reinforcing the need for medication therapy monitoring and management.
Resources for Pharmacists: Information about XLH for patients and health professionals is available online at the XLH Network
and XLH Link.
References
- Manghat P, Sodi R, Swaminathan R. Phosphate homeostasis and disorders. Ann Clin Biochem. 2014;51(Pt 6):631–656.
- Carpenter TO, Imel EA, Holm IA et al. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26:1381–1388.
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131–155.
- Pai AB. Disorders of calcium and phosphorus homeostasis. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey LM, eds. Pharmacotherapy: A Pathophysiologic Approach. 10th ed. New York, NY: McGraw-Hill; 2017.https://accesspharmacy.mhmedical.com/content.aspx?sectionid=146061894&bookid=1861&jumpsectionID=146062030&Resultclick=2#1148574116. Accessed August 9, 2018.
- Cross HS, Debiec H, Peterlik M. Mechanism and regulation of intestinal phosphate absorption. Miner Electrolyte Metab. 1990;16(2-3):115–124.
- Razzaque MS. The FGF23–klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5(11):611–619.
- XLH Network. What Is XLH? August 13, 2017. http://xlhnetwork.org/what-is-xlh/symptoms-of-xlh. Accessed July 22, 2018.
- Sharkey MS, Grunseich K, Carpenter TO. Contemporary medical and surgical management of X-linked hypophosphatemic rickets. J Am Acad Orthop Surg. 2015;23(7):433–442.
- Carpenter TO, Whyte MP, Imel EA et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018; 378:1987–1998.
- Nield LS, Mahajan P, Joshi A et al. Rickets: not a disease of the past. Am Fam Phys. 2006;74:619–630.
- Ruppe MD. X-linked hypophosphatemia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews [Internet]-NCBI Bookshelf. Seattle, WA: University of Washington; 1993–2017.
- Linglart A, Biosse-Duplan M, Briot K et al. Therapuetic management of hypophosphatemia rickets from infancy to adulthood. Endocrine Connections. 2014;3:R13–R30.
- Vega RA, Opalak C, Harshbarger RJ et al. Hypophosphatemic rickets and craniosynostosis: a multicenter case series. J Neurosurg Pediatr. 2016;17:694–700.
- Kawaguchi A, Miyamoto K, Wakahara K et al. Surgical treatment of multiple spinal canal stenosis associated with vitamin D-resistant rickets. J Clin Neurosci. 2009;16:717–719.
- Glorieux FH, Marie JP, Pettifor JM, et al: Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin-D resistant rickets. N Engl J Med. 1980;303:1023–1031.
- Scheinman SJ, Drezner MK. Hereditary Hypophosphatemic Rickets and Tumor-Induced Osteomalacia. UpToDate. December 1, 2015; http://www.uptodate.com/contents/hereditary-hypophosphatemic-rickets-and-tumor-induced-osteomalacia?source=search_result&search=x-linked+hypophosphatemia&selectedTitle=1~10. Accessed July 22, 2018.
- Pavone V, Testa G, Iachino SG et al. Hypophosphatemia rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol. 2015;25:221–226.
- Crysvita (burosumab-twza injection) prescribing information. Novato, CA: Ultragenyx Pharmaceuticals Inc; 2018.
- Imel EA, Zhang X, Ruppe MD, et al. Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab. 2015;100(7):2565–2573.
- Lamb YN. Burosumab: first global approval. Drugs. 2018;78:707–714.
Back to Top