
Expired activity
Please go to the PowerPak
homepage and select a course.
The Impacts of Biosimilars on Pharmacists: Implications of the Evidence on Treatment Choices
INTRODUCTION
Biologic products (biologics) are complex molecules derived from living cells that are cultured, manufactured, and packaged in formulations used for the treatment, prevention, or cure of disease in humans [US FDA “Frequently asked questions;” US FDA “What are biologics”]. Since the first biologic, diphtheria vaccine isolated from the blood of horses, was developed at the end of the 19th century, biologics have revolutionized the treatment of many serious medical conditions, including anemia, diabetes mellitus, hepatitis, multiple sclerosis, inflammatory bowel disease, rheumatoid arthritis, and cancer [APA March 6, 2015; Bristow 2006; Pharmacy Practice News May 2013; US FDA “Information for consumers;” US FDA “What are biologics”]. However, these clinical benefits come at significant costs to the healthcare system because these complex molecules are costly to successfully develop and commercialize for therapeutic use [Lucio 2013; Schumock 2015]. The cost of biologics also results in limited access for many patients who could benefit [Wolfe 2009].
In order to drive down costs and increase access to biologics, the United States (US) Food and Drug Administration (FDA) was given authority to approve “lower cost, follow-on versions of previously approved biologics,” or biosimilars, under the Affordable Care Act (ACA) [Lucio 2013]. The first biosimilar, filgrastim-sndz, a biosimilar of filgrastim was approved by the US FDA in March 2015 and was launched in September 2015 [US FDA March 6, 2015; Reuters Sept 3 2015]. Since the manufacturing process for biosimilars is complex, there are concerns about their efficacy and safety. As medication therapy experts, pharmacists will play a major role in educating other healthcare professionals about biosimilars, developing recommendations and formulary reviews, and ensuring these agents are incorporated into clinical practice to maximize the opportunities for patients to benefit [Lucio 2013; APA Jan 1, 2015].
After completing this activity, pharmacists will be able to:
- Identify the key differences between biosimilars, biologics, and generics as pertaining to production, regulatory requirements, and costs
- Evaluate the rationale for the use of biosimilars in patient care
- Assess the evidence of biosimilar agents for oncology, hematology, and inflammatory disease
- Review current United States (US) and out-of-US experiences with biosimilars
What Are Biosimilars? And Why Are They Important?
Since the introduction of recombinant human insulin in 1982, the number of biologics available for therapeutic use has increased dramatically (Figure 1) [Lucio 2013; Kling 2014]. There are currently more than 100 biologics available for therapeutic use in the US, and many more are in development. Specific examples and their indications include [Pharmacy Practice News May 2013]:
- Basiliximab (monoclonal antibody indicated for prophylaxis of acute organ rejection in renal transplant)
- Albumin (hypovolemia)
- Interferon alfa-2B (melanoma)
- Epoetin alfa (anemia due to chronic kidney disease)
- Recombinant human insulin and pegloticase (refractory chronic gout)
- Rituximab (lymphoma)
| Figure 1: Approval of FDA new molecular entities and biological license approvals 1998–2013. [Kling 2014]. |
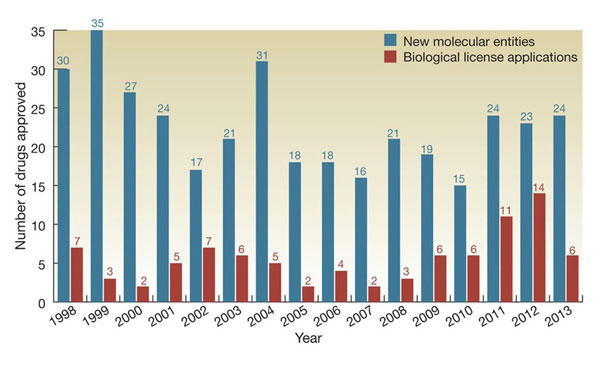 |
US FDA Definition of Biological (Biologic):
“Biological product” means:
- A virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, or analogous product, or arsphenamine or derivative of arsphenamine (or any other trivalent organic arsenic compound)
- Applicable to the prevention, treatment, or cure of a disease or condition of human beings [Public Health Service Act Section 351(i)]
- Biological products also meet the definition of either a drug or device under Sections 201(g) and (h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act).
|
| Available at: http://www.fda.gov/ICECI/Inspections/IOM/ucm122535.htm. Accessed January 14, 2015. |
Biologics provide novel and effective treatment options for patients with many common and/or serious medical conditions. In 2009, global sales of biologics were $93 billion [Lucio 2013]. In a 2009 publication, it was projected that by 2016, 10 of the top 20 selling drugs globally would be biologics [McCamish 2011; Lucio 2013]. And this prediction will likely come true – based on 2014 data, 8 of the top drugs by expenditure in nonfederal hospitals were biologics (infliximab, rituximab, pegfilgrastim, immune globulin, alteplase, natalizumab, bevacizumab, trastuzumab) [Schumock 2015]. Biologics contributed significantly to an overall 12.4% annual growth rate in drug expenditures from 2013 to 2014 (Figure 2) [Schumock 2015].
| Figure 2. Total US Drug and Biologic Expenditures [Schumock 2015]. |
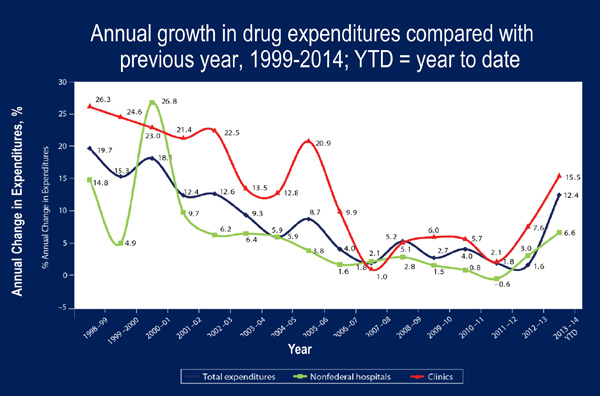 |
The Biologics Price Competition and Innovation (BPCI) Act of 2009, which is under the umbrella of the Affordable Care Act, created a new pathway for the US FDA to approve “copies” of innovator biologics, or “biosimilars.” The major goals of the BPCI Act are to decrease costs and increase access to biologic therapies [Blackstone 2013; Palmisano 2012; Johnson 2015]. Third-party payers anticipate that biosimilars will cost 40% to 50% less than originator brands; however, this may vary depending on initial introductions of biosimilars into the US healthcare market [Reuters Sept 3 2015]. Express Scripts estimates that $250 billion could be saved between 2014 and 2024 if only 11 of the most likely to be approved biosimilars enter the market [Express Scripts 2013]. Increased competition among biologics manufacturers should also lead to increased innovation, and ultimately to more treatment options for patients [Blackstone 2013].
The early impact of biosimilars in the US market will soon become apparent. Filgrastim-sndz, a biosimilar of filgrastim, was approved by the US FDA in March 2015, and was launched in September 2015 after a delay caused by appeals from the manufacturer of the originator product (filgrastim) [US FDA March 6, 2015; Reuters Sept 3 2015]. Data from IMS Health indicate that use of another filgrastim (tbo-filgrastim) resulted in cumulative cost savings in hospitals of almost $14,000,000 [IMS Health]. An application for a biosimilar of infliximab is currently being reviewed by the US FDA and approval is anticipated in the near future [Biopharma May 26, 2015]. Many other biosimilars are currently in phase I and II trials and are expected to be introduced to the US market over the next five years (Figure 3) [APA Jan 1, 2015; IMS Health].
| Figure 3: Growth Potential of the US Biosimilars Market [Adapted from IMS Health]. |
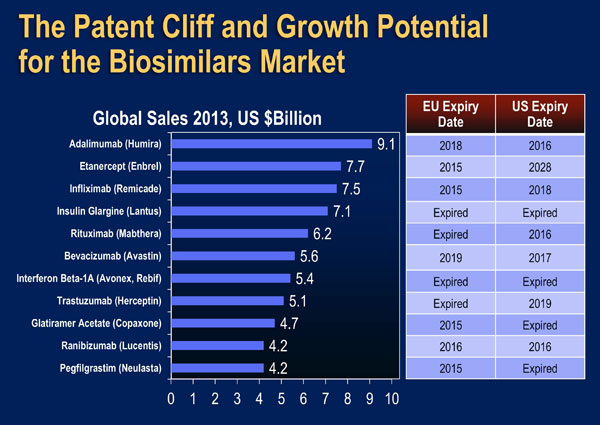 |
However, manufacturing and marketing biosimilars is “easier said than done.” Biologics are large (typically up to 150,000 daltons and containing 20,000 atoms) and heterogeneous molecules with complex structures. They have structurally specific and targeted mechanisms of action based on binding to multiple cell surface receptors. Because of their inherent complexity, biologics are produced using biotechnology and other cutting-edge methods. Biologic molecules also tend to degrade over time, so they have special packaging and storage requirements. As described in detail below, even a small change in any step in the production or formulation processes may lead to important changes to the molecule. Therefore, exact copies of biologics, whether innovator products or follow-on biosimilars, cannot be made [Schellekens 2009; GaBI 2012; DeClerck 2012].
In contrast, traditional generics are small molecule drugs (SMDs) of low molecular weight molecules and with fairly simple manufacturing process [Pharmacy Practice News May 2013; DeClerck 2012]. Therefore, generic version of SMDs can be considered equivalent to the original compound. Figure 4 illustrates the comparison between a biologic and an SMD. Table 1 summarizes the major differences between biologicals and SMDs. These differences lead to important considerations when evaluating the biologic and clinical comparability of biosimilars [GaBI 2012].
| Figure 4: Comparison between a Biologic (Human Erythropoietin) and a Small Molecule Drug (Cisplatin) [Illustration courtesy of: Olgun Guvench, MD, PhD; University of New England College of Pharmacy.] |
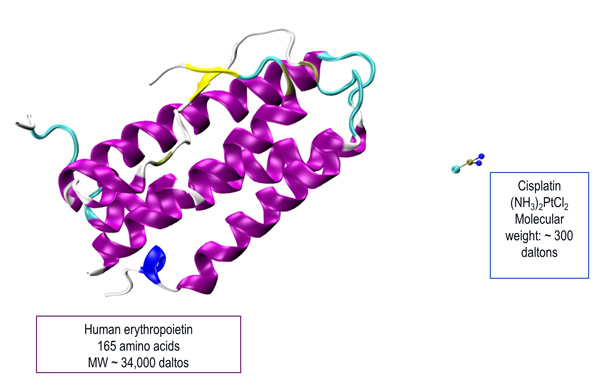 |
| Table 1: Characteristics of Small Molecule Drugs Compared to Biologics [GaBI 2012]. |
| |
Small Molecule Drugs |
Biologics |
| Size |
- Small (single molecule)
- Low molecular weight
|
- Large (mixture of related molecules)
- High molecular weight
|
| Structure |
Simple, well-defined, independent of manufacturing process |
Complex (heterogeneous), defined by the exact manufacturing process |
| Post-translational Modification |
None |
Several types (i.e. glycosylation, methylation, phosphorylation) |
| Manufacturing |
- Produced by chemical synthesis
- Predictable chemical process
- Identical copy can be made
|
- Produced in living cell culture
- Difficult to control from starting material to final product
- Impossible to ensure identical copy
|
| Characterization |
Easy to characterize completely |
Cannot be characterized completely due to the molecular complexity and heterogeneity |
| Stability |
Stable |
Unstable, sensitive to external conditions |
| Immunogenicity |
Non-immunogenic |
Immunogenic |
See comment in PubMed Commons below Because biologics are proteins derived from living cells, the manufacturing process is complex, and involves various biotechnological processes, such as recombinant DNA technology, controlled gene expression, and antibody methods [Pharmacy Practice News May 2013]. As shown in Figure 5, key steps in the manufacturing process for manufacturing a recombinant human protein include cloning of the protein into a DNA vector, transfer into a host cell for amplified expression, cell expansion and cell production, recovery of the protein of interest, purification of the molecule, and formulation of the bulk medication [Pharmacy Practice News May 2013; Mellstedt 2008]. Each of the steps in the process has some inherent variability, especially because living organisms are involved and the details of the processes developed by different companies are proprietary. [Kuhlmann 2010].
| Figure 5: Steps in Recombinant Protein Production [Adapted from Mellstedt 2008] |
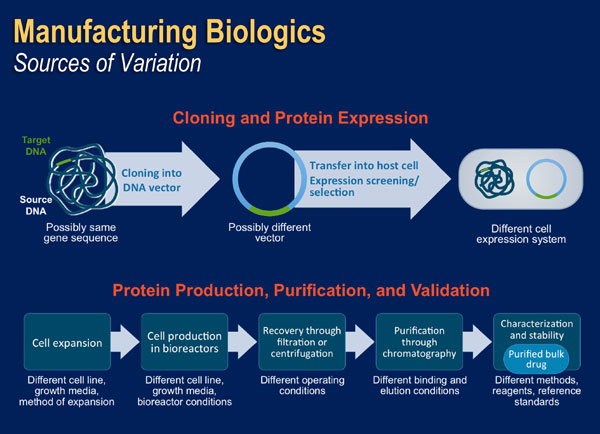 |
When any of the key steps involved in manufacturing a biologic are changed, the quality and function of the final product may be altered [Jelkmann 2007]. Even small changes in, or differences between, manufacturing processes may have a significant impact on the quality, purity, biological characteristics, and clinical activity of the final product. This is true for any biological product including innovator products and biosimilars. Changes may occur to the expression systems used for production, culture conditions (e.g. temperature, pH, and nutrients), purification, processing, and formulation. These differences may lead to structural differences between proteins, including modification of the protein primary sequence, glycosylation patterns, or the conformational state of the molecule. In addition, conditions for storage (e.g. light and temperature) and packaging may also affect the stability of the final product.
Even if biosimilars are produced from the same recombinant DNA using the same technique, formulation, and packaging as the innovator product, there may be differences with the final product as compared with the reference product [Schellekens 2009; Mikhail 2013]. The US FDA Guidance for Industry on Biologics notes that: “The demonstration of comparability does not necessarily mean that the quality attributes of the pre-change and post-change product are identical, but that they are highly similar and that the existing knowledge is sufficiently predictive to ensure that any differences in quality attributes have no adverse impact upon safety or efficacy of the drug product.” [US FDA June 2005].
Because many biosimilars are expected to be approved in the US in the next few years (Figure 3), pharmacists must have a good understanding of the manufacturing process for biologics and the potential for differences in pharmacokinetics, pharmacodynamics, and clinical parameters. In addition, pharmacists must understand how the US FDA’s review, approval, and pharmacovigilance standards are designed to ensure safe and effective use of biologics [Brinks 2011; Lucio 2013; Nascimento 2015].
The Potential for Immunogenicity
One of the major concerns about biosimilars is the potential for immunogenicity, or the potential to provoke an immune response with the potential for adverse therapeutic consequences, such as formation of neutralizing antibodies to the therapeutic protein or cytokine release leading to hypersensitivity. All biologics confer a risk of immunogenicity that is related to patient, disease, and product factors. Changes to the structure of the protein molecule may increase the potential for immunogenicity – this may be seen with lot-to-lot variability for a single manufacturer, as well as among different manufacturers. Because of the potential for adverse clinical consequences, such as diminished efficacy or adverse events, ongoing monitoring of biologic agents is important and is a key component of pharmacovigilance [US FDA Dec 2012].
The case of Eprex®, an epoetin-alfa originator product marketed outside the US by Johnson and Johnson, highlights the need for post-marketing surveillance for biologics. Coincidentally with the availability of Eprex®, there was a large increase in the incidence of antibody-mediated pure red cell aplasia (PRCA) in patients with chronic kidney disease (CKD) treated with the product [Schellekens 2009]. PRCA is a potentially fatal condition in which anti-epoetin antibodies neutralize both the exogenously administered medication and endogenous erythropoietin, leading to severe and intractable anemia [Kuhlmann 2010]. Before Eprex® was marketed in 1998, only three cases of PRCA had been reported. However, the incidence in patients with CKD increased sharply to about 250 cases between 1998 and 2002. Many patients became completely transfusion dependent, and the mortality rate was high [Boven 2005; Locatelli 2007; Kuhlmann 2010]. The events were associated most commonly with subcutaneous rather than intravenous injection.
Variation in the structure of the epoetin molecule itself was ruled out as a contributing factor. The increase in immunogenicity and resulting PRCA events coincided with a relatively minor formulation change, the replacement of human serum albumin as a stabilizer with glycine and polysorbate 80 in 1998. This might have promoted the formation of epoetin-containing micelles that the body’s immune system saw as foreign pathogens, or the polysorbate 80 may have reacted with the uncoated rubber stoppers of the prefilled syringes to produce leachates that provoked an antibody response [Kuhlmann 2010]. One additional possibility is that the new formulation was less stable and, therefore, more susceptible to denaturation or aggregation under inadequate storage or improper handling conditions [Kuhlmann 2010].
In 2003, the uncoated rubber stoppers in the prefilled syringes were replaced with teflon-coated stoppers, enhanced control of the product’s cold chain was implemented, and detailed guidelines emphasizing the importance of storage at between 2 and 8 degrees Celsius were implemented. In addition, intravenous administration was recommended over subcutaneous administration. As a result of these changes, the incidence of PRCA decreased dramatically to a level similar to that seen prior to 1998. In order to mitigate the risks of immunogenicity and other potential adverse clincal effects, changes in the manufacturing process of biologics for each manufacturer are highly regulated by the FDA and the European Medicines Agency (EMA) [Kuhlmann 2010].
The Standards for Approval of Biosimilars: Focus on Patient Access and Safety
The US FDA has established a rigorous review and approval process for biosimilars that is, in large part, based on experience gained in the EU, where biosimilars have been available since 2006. EMA was a leader in establishing an overarching guideline that defines biosimilars and establishes a process for demonstrating similarity between a biosimilar and the reference innovator product. The EMA guideline is based on demonstrating comparability based on quality, efficacy, clinical efficacy, and clinical safety.
Similarly, the FDA emphasizes that a “totality of the evidence” approach will be used in reviewing all of the information submitted in support of biosimilar approval, including detailed analytical testing, clinical immunogenicity evaluation, animal studies, and human clinical trials. Comparator data involving a non-US-licensed product in support of the overall biosimilar application may be used in addition to other evidence [Lucio 2013; US FDA June 2005]. The goal of the biosimilarity review is to establish that the candidate biosimilar is not significantly different from the reference product and is unlikely to have any clinically significant differences. In general, smaller-scale direct comparisons and extrapolation are used [US FDA June 2005].
The FDA will allow the biosimilar sponsor to pursue all or only a subset of indications and routes of administration of the innovator product. However, this allowance does not eliminate the need for clinical data comparing the proposed biosimilar with the originator’s US-licensed product [Lucio 2013].
Regulatory Definitions of a Biosimilar
Food and Drug Administration (USA)
A biological product that is highly similar to a US-licensed reference biological product notwithstanding minor differences in inactive components and for which there are no clinically meaningful differences in safety, purity, or potency of the product
European Medicines Agency – Europe
…structurally highly similar versions of an already authorized biological medicinal product (the reference product) with demonstrated similarity in physicochemical characteristics, efficacy, and safety based on a comprehensive comparability exercise
|
FDA website:
http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf.
Weise M et al. Nat Biotechnol. 2011;29:690-693. |
The US FDA encourages biosimilar applicant companies to use a “step-wise” process for biosimilar development (Figure 6). At each step, the biosimilar applicant should identify the extent of remaining uncertainty regarding the demonstration of biosimilarity, and use subsequent development steps to address any residual concerns (Figure 7).
| Figure 6: A Stepwise approach to demonstrating biosimilarity [Adapted from McCamish 2012] |
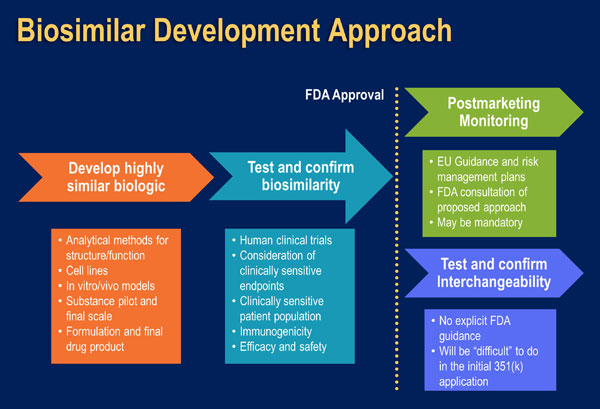 |
| Figure 7: Assessments of Analytical Characterization and Residual Uncertainty [US FDA April 2015]. |
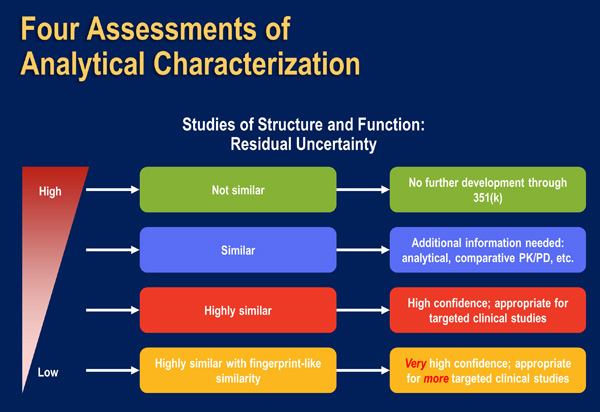 |
Companies submitting must be able to demonstrate the comparability between their proposed product and the innovator reference product based on [Lucio 2013]:
- primary structure (i.e. amino acid sequence) and higher order structures
- function (demonstration that the mechanism of action is the same)
- enzymatic post-translational modifications (i.e. glycosylation, methylation, phosphorylation)
- animal toxicity studies
- clinical immunogenicity studies
- clinical studies
The structure and function evaluations will serve as the foundation for biosimilar development and results are useful to determine what additional studies are necessary. The capability of the methods used in the analytical assessments, as well as their limitations, must be adequately and comprehensively described by the company submitting the biosimilar for review and approval. Physicochemical and functional characterization studies should be sufficient to establish relevant quality attributes, including those that define a product’s identity, quantity, safety, purity, and potency [US FDA June 2005]. Human pharmacokinetic studies in clinically relevant patient populations should include measures that can be quickly assessed with precision and that are sensitive enough to detect clinically meaningful differences. It is recommended that studies will utilize crossover and parallel designs and correlate exposure with clinical outcomes. Comparative parallel clinical immunogenicity studies are also important to evaluate potential differences in the incidence and severity of immune responses using endpoints such as antibody formation (binding, neutralizing) and cytokine levels; as noted, immunogenicity is also an important component of pharmacovigilance [US FDA June 2005].
Clinical studies may be required in populations with the medical condition(s) for the target indication(s) and with clinically relevant endpoints that are sensitive to detecting meaningful differences in safety and efficacy depending on what questions remain in these areas. The biosimilar applicant company will be required to justify any differences in comparability [Lucio 2013; Schellekens 2009]. FDA scientists will evaluate the applicant’s integration of various types of information to provide advice on the scope and extent of the development plan and, ultimately, an overall assessment that a biological product is (or is not) a biosimilar to an approved reference product [Lucio 2013; US FDA June 2005].
A biosimilar company may seek approval of their product in one of two categories based on the BICI Act:
- “Biosimilar” if data show that the product is “’highly similar’ to the reference product, notwithstanding minor differences in clinically inactive components, and that no clinically meaningful differences exist between the biological product and the reference product in terms of safety, purity, and potency.
- “Interchangeable” if the sponsor demonstrates “that the biosimilar product can be expected to produce the same clinical result as the reference product in any given patient and, for a biological product that is administered more than once, that the risk of safety or diminished efficacy of alternating or switching between use of the biosimilar product and the reference product is not greater than the risk of maintaining the patient on the reference product. Interchangeable products may be substituted for the reference product by a pharmacist without the intervention of the prescribing healthcare provider, as allowed by state laws and regulations.”
The first biosimilar approved by the US FDA, filgrastim-sndz was approved with the “biosimilar” designation. The submitting company did not seek the “interchangeable” designation because the US FDA has not yet clearly defined the requirements for this designation [MacDonald 2015]. The potential for “therapeutic substitution” of a biologic for an innovator product is also a source of great concern among prescribers and will have to be addressed as the US biosimilar approval process evolves. As the key institutional and clinical decision-makers in the case of therapeutic substitution, clinical pharmacists must take a leadership role to ensure that biosimilars are optimally incorporated into clinical practice [Schellekens 2009; Lucio 2013].
The regulatory requirements for approval of biosimilars “are stringent and require thorough evaluation of safety and efficacy” [Lucio 2013]. However, the process is abbreviated compared to innovator products and the pool of patients and clinical data is smaller. Therefore, even with data from clinical trials, rare adverse events unique to the biosimilar may not be apparent prior to marketing. Pharmacists must take an active role to ensure that pharmacovigilance programs are in place to ensure the safe and effective use of these agents in clinical practice. Key responsibilities in promoting biosimilar pharmacovigilance programs include tracking administration of specific products using bar codes, maintenance of electronic medical records, and medication reconciliation [Zuñiga 2010; Casadevall 2013; Felix 2014].
In summary, the era of biosimilars is now a reality in the US. As these agents are introduced to the market, pharmacists must take an active role to educate colleagues about their review and approval process, and ensure they are appropriately integrated into formularies and clinical guidelines. Although traditional generic drugs deemed bioequivalent by the US FDA do not generally require a review by institutional Pharmacy and Therapeutics (P&T) committees, biosimilars may vary in terms of labeled indications and naming. Therefore pharmacists must lead development of policies and procedures to guide these reviews. They must also be involved in decisions regarding the evolving issues of interchangeability and development and implementation of pharmacovigilance programs [Lucio 2013]. Pharmacists should also take an active role to facilitate transitions of care for patients who have recently been prescribed a biosimilar medication [Zuñiga 2010; Casadevall 2013; Felix 2014]. By taking a leadership role, pharmacists can help ensure that biosimilars achieve the goals of providing effective treatment options to more patients, reducing healthcare costs, and spurring healthcare innovation.
Evaluating Data and Incorporating Biosimilars in Clinical Practice – Lessons from the European Experience
Important insights can be gained from the experience with biosimilars in Europe, where there are almost 20 agents currently available (Table 2).
| Table 2: Biosimilars Available in Europe [EMA 2015]. |
| European Biosimilars Experience |
| Active Substance |
Products |
Approval |
| Epoetin alfa |
Abseamed
Binocrit
Epoetin Alfa Hexal |
8/2007
8/2007
8/2007 |
| Epoetin zeta |
Retacrit
Silapo |
12/2007
12/2007 |
| Filgrastim |
Accofil
Biograstim
Filgrastim Hexal
Grastofil
Nivestim
Ratiograstim
Tevagrastim
Zarzio |
9/2014
9/2008
2/2009
10/2013
6/2010
9/2008
9/2008
2/2009 |
| Follitropin alfa |
Bemfola
Ovaleap |
3/2014
9/2013 |
| Infliximab |
Inflectra
Remsima |
9/2013
9/2013 |
| Insulin glargine |
Abasaglar/Abasria |
9/2014 |
| Somatropin |
Omnitrope |
4/2006 |
Two biosimilars (Remsima®, Inflectra®) of the innovator infliximab (Remicade®) were approved by the EMA in 2013. Both biosimilars are derived from the same type of cell line and have the same amino acid sequence as the innovator infliximab, although the exact cell line used is specific to each biosimilar manufacturer. Based on a comprehensive comparative program, including two pivotal randomized controlled trials (RCTs) conducted in patients with rheumatoid arthritis (PLANTERA [Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in RA patients]) and ankylosing spondylitis (PLANETAS [Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in AS patients]), CT-P13 was approved by the EMA across all indications for which the infliximab innovator product was approved, including inflammatory bowel disease (IBD), Crohn’s disease (CD), and ulcerative colitis (UC) [Jung 2014; De Groot 2007].
Extensive analytical tests and clinical studies between the two biosimilars and the innovator infliximab showed comparable [GaBI 2015; Park 2013; Yoo 2013; Yoo 2015]:
- physicochemical and structural comparability (except for a small difference in the proportion of fucosylated forms)
- receptor binding
- mechanism of action (inhibition of direct effects of TNFα on epithelial cells and induction of regulatory macrophages)
- pharmacokinetics
- efficacy based on the American College of Rheumatology ACR20 response at week 30 in the RA study and the Assessments in Ankylosing Spondylitis ASAS20 response
- safety
- proportion of patients with anti-drug antibodies at weeks 14, 30, and 54
- incidence of serious infections, malignancy and lymphoma, and infusion-related reactions
A study of 39 RA and AS patients who were switched from the innovator product to the CT-P13 biosimilar infliximab after a mean of 4.1 years of treatment showed that symptom levels, disease activity, and effectiveness were similar over a follow-up of 11 months. No immediate safety signals were observed [Sokka 2015; GaBI 2015].
Pharmacovigiliance Example from the EU
Four biosimilars of the innovator product of granulocyte colony stimulating factor (G-CSF, Neupogen) have been available in Europe since 2008 and are now in widespread clinical use: Zarzio/Filgrastim Hexal, Tevagrastim/Ratiograstim, Nivestim, and Grastofil [Bonig 2015]. One of these biosimilars (Zarzio®) launched in 2009 and has an estimated exposure of approximately 4.5 million patient days, as an example of the pharmacovigilance studies to which all biosimilars are subjected. A pooled analysis of 5 post-approval studies of Zarzio® included 1,302 adult patients who were treated with at least one cycle of chemotherapy and G-CSF support for the prevention of neutropenia. The occurrence of severe or febrile neutropenia was within the range of that observed in previous studies of originator G-CSF [Gascón 2013]. Another international, observational, open-label, pharmacoepidemiologic study (MONITOR-GCSF) of cancer patients (n = 1447) treated with myelosuppressive chemotherapy who are receiving prophylaxis with Zarzio® is ongoing in 12 countries to evaluate real-world treatment patterns and outcomes. See comment in PubMed Commons below Based on results reported in 2015, clinical and safety outcomes were well within the range of historically reported data for originator filgrastim. This reinforces the clinical effectiveness and safety of biosimilar filgrastim in daily clinical practice [Gascón 2013].
SUMMARY
New biosimilar medications across multiple therapeutic areas are expected to be approved in the US in the next few years. As these agents are introduced into clinical practice and the FDA’s guidance regarding review, approval, and ongoing monitoring evolves, pharmacists must keep up to date with current information in order to educate other healthcare professionals and ensure that processes are in place to confirm the effective and safe use of biosimilars [Grampp 2015]. In the meantime, it is helpful to review and apply lessons from the extensive biosimilar experience gained in the European market.
In addition, as key members of the clinical care team, pharmacists can educate other healthcare practitioners about important factors to consider when evaluating biosimilars [Mikhail 2013]:
- Pre-registration clinical trials, study design, sample size
- Study population and how representative it is of the clinical population
- Study duration, statistical methodology
- Difference between the trial and innovator product (biologic activity, route of administration, median dosage, endpoints)
- The need to establish local protocols/care bundles to avoid inadvertent interchange or switching administration route (IV vs SC)
- Safety, adverse events, potential for immunogenicity
- Post-marketing data, clinical experience, adverse event reporting
REFERENCES
- APA (American Pharmacists Association). Pharmacists and health systems prepare to add biosimilars to formularies. Jan 1, 2015. Available at: https://www.pharmacist.com/pharmacists-and-health-systems-prepare-add-biosimilars-formularies. Accessed Nov 20, 2015.
- APA (American Pharmacists Association). FDA approves first biosimilar drug. March 6, 2015. Available at: https://www.pharmacist.com/fda-approves-first-biosimilar-drug. Accessed Nov 20, 2015.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- BioPharma. Remsima’s US FDA review held back by contradictory demands, says Celltrion. May 26, 2015. Available at: http://www.biopharma-reporter.com/Markets-Regulations/Remsima-s-US-FDA-review-held-back-by-data-demands-says-Celltrion. Accessed Nov 30, 2015.
- Bonig H et al. Biosimilar granulocyte–colony-stimulating factor for healthy donor stem cell mobilization: need we be afraid? Transfusion. 2015;55.2:430-439.
- Boven K, Knight J, Bader F et al. Epoetin-associated pure red cell aplasia in patients with chronic kidney disease: solving the mystery. Nephrol Dial Transplant. 2005;20 Suppl 3:iii33-40.
- Brinks V, Hawe A, Basmeleh AF et al. Quality of original and biosimilar epoetin products. Pharm Res. 2011;28:386-393.
- Bristow AF, Barrowcliffe T, Bangham DR. Standardization of biological medicines: the first hundred years, 1900–2000. Notes Rec R Soc. 2006;60:271-289.
- Casadevall N, Edwards IR, Felix T et al. Pharmacovigilance and biosimilars: considerations, needs and challenges. Expert Opin Biol Ther. 2013;13:1039-1047.
- DeClerck PJ. Biosimilars: demonstrating a ‘similarity.’ February 17, 2012. Available at: http://gabionline.net/layout/set/print/content/view/full/1724. Accessed Dec 8, 2015.
- De Groot AS, Scott DW. Immonogenicity of protein therapeutics. Trends Immunol. 2007;28:482-490.
- EMA (European Medicines Agency) website. Available at: http://www.ema.europa.eu. Accessed June 2015.
- Express Scripts. The $250 billion potential of biosimilars. April 23, 2013. Available at: http://lab.express-scripts.com/insights/industry-updates/the-$250-billion-potential-of-biosimilars. Accessed Jan 10, 2016.
- Felix T, Johansson TT, Colliatie JA et al. Biologic product identification and US pharmacovigilance in the biosimilars era.Nat Biotechnol. 2014;32:128-130.
- GaBI (Genetics and Biosimilars Initiative). Small molecule versus biological drugs. June 29, 2012. Available at: http://www.gabionline.net/Biosimilars/Research/Small-molecule-versus-biological-drugs. Accessed Nov 17, 2015.
- GaBI (Genetics and Biosimilars Initiative). Extrapolation of indications in biosimilars: infliximab. Sept 1, 2015. Available at: http://gabionline.net/Biosimilars/Research/Extrapolation-of-indications-in-biosimilars-infliximab. Accessed Dec 8, 2015.
- Gascón P, Tesch H, Verpoort et al. Clinical experience with Zarzio® in Europe: What have we learned? Support Care Cancer. 2013;21:2925-2932.
- Grampp G, Thomas F. Pharmacovigilance considerations for biosimilars in the USA. Biodrugs. 2015;29.5:309-321.
- IMS Health. Data on file. Also, available at: http://www.imshealth.com/en. Accessed June 2015.
- Jelkmann W. Recombinant EPO production—points the nephrologist should know. Nephrol Dial Transplant. 2007;22:2749-2753.
- Johnson SR. FDA panel recommends first biosimilar approval. Jan 7, 2015. Available at: http://www.modernhealthcare.com/article/20150107/NEWS/301079947. Accessed Nov 17, 2015.
- Jung SK, Lee KH, Jeon JW et al. Physiochemical characterization of Remsima. Mabs. 2014;6:1163-1177.
- Kuhlmann M, Marre M. Lessons learned from biosimilar epoetins and insulins. Achieving Best Practice. 2010;90-97.
- Kling J. Fresh from the biotech pipeline — 2013. Nat Biotechnol. 2014;32:121-124.
- Locatelli F, Goldsmith D. Biosimilars: Uncharted territory. Perit Dial Int. 2007;27:S303-S307.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- MacDonald G. Biosimilar not Interchangeable: Sandoz and Pfenex call for US FDA Guidelines. Sept 4, 2015. Available at: http://www.biopharma-reporter.com/Markets-Regulations/Biosimilar-not-interchangeable-Sandoz-and-Pfenex-call-for-US-FDA-guidelines. Accessed Dec. 8, 2015.
- McCamish M, Woollett G. Worldwide experience with biosimilar development. mAbs. 2011;3:209-217.
- McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91:405-417.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19:411-419.
- Mikhail A, Farouk M. Epoetin biosimilars in Europe: five years on. Adv Ther. 2013;30:28-40.
- Nascimento MC, Abreu CLC, Cosata RN et al. Braz Arch Biol Technol. 2015;1-7.
- Palmisano DJ. Patient safety top concern with biosimilars. Oct 15, 2012. Available at: http://www.genengnews.com/gen-articles/patient-safety-top-concern-with-biosimilars/4555. Accessed Nov 17, 2015.
- Park W, Hrycaj P, Jeka S et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Pharmacy Practice News. Understanding key differences between biosimilars and small molecule generics. Special report: May 2013. Available at: http://www.pharmacypracticenews.com/download/sr1229_wm.pdf. Accessed Nov 17, 2015.
- Reuters; Sept 3, 2015. Novartis launches first U.S. ‘biosimilar’ drug at 15 percent discount. Available at: http://www.reuters.com/article/2015/09/03/us-novartis-drug-idUSKCN0R30C220150903#ufxrhbHicQZxPGpz.97. Accessed Nov 20, 2015.
- Schellekens H. Biosimilar therapeutics – what do we need to consider? NDT Plus. 2009;2(suppl 1):i27-i36.
- Schumock GT, Li EC, Suda KJ et al. National trends in prescription drug expenditures and projections for 2015. Am J Health Syst Pharm. 2015;72:717-736.
- Sokka T, Kautiainen H. SAT0174 clinical experience with infliximab biosimilar – switch from remicade. Ann Rheum Dis. 2015;74:717.
- US FDA (United States Food and Drug Administration). FDA approves first biosimilar product Zarxio. March 6, 2015. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. Accessed Nov 20, 2015.
- US FDA (United States Food and Drug Administration). Frequently asked questions about therapeutic biological products. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/%20HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm113522.htm. Accessed Nov 1, 2015.
- US FDA (United States Food and Drug Administration). Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. April 2015. Available at: http://www.fda.gov/downloads/DrugsGuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed Jan 26, 2016.
- US FDA (United States Food and Drug Administration). Guidance for industry: Q5E comparability of biotechnological/biological products subject to changes in their manufacturing process. June 2005. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073476.pdf. Accessed Nov 10, 2015.
- US FDA (United States Food and Drug Administration). What are biologics? Available at: http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CBER/ucm133077.htm. Accessed Nov 15, 2015.
- US FDA (United States Food and Drug Administration). Information for consumers (biosimilars). Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm241718.htm. Accessed Nov 17, 2015.
- US FDA (United States Food and Drug Administration). Guidance for Industry and Investigators: Safety Reporting Requirements for INDs and BA/BE Studies. Dec 2012. Available at: http://www.fda.gov/downloads/Drugs/.../Guidances/UCM227351.pdf.
- Wolfe F, Michaud K. Out-of-pocket expenses and their burden in patients with rheumatoid arthritis. Arthritis Rheum. 2009;61:1563-1570.
- Yoo DH, Hrycaj P, Jeka S et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1613-1620.
- Yoo DH, Oh C, Hong S, Park W. Analysis of clinical trials of biosimilar infliximab (CT-P13) and comparison against historical clinical studies with the infliximab reference medicinal product. Expert Rev Clin Immunol. 2015;11(supp 1):15-24.
- Zuñiga L, Calvo B. Biosimilars: Pharmacovigilance and risk management. Pharmacoepidemiol Drug Saf. 2010;19:661-669.
Back to Top