
Expired activity
Please go to the PowerPak
homepage and select a course.
Defining the Role of the Pharmacist in the Management of Von Willebrand Disease: Improving Patient Outcomes
INTRODUCTION
Von Willebrand disease (VWD) is the most common type of bleeding disorder. It is usually an inherited condition, though it may be acquired, caused by a missing or a defective von Willebrand factor (VWF),1 which is an elongated, multimeric protein that is required for normal adhesion of platelets at the site of a blood vessel injury and for protection of factor VIII (FVIII) in circulation. Thus, when VWF levels are reduced or malfunctioning, a patient is more likely to bleed.2 Most patients with VWD suffer from mucosal bleeding and are more likely to bleed after surgery and trauma than patients without bleeding disorders.2 Some patients with severe forms of VWD suffer from gastrointestinal or joint bleeding.2 Several different types of VWD exist, and the type determines the best course of treatment, but the overall treatment approach for VWD is based on normalizing VWF and FVIII levels. This review will describe the etiology, epidemiology, diagnosis, and treatment options for patients with VWD.
ETIOLOGY OF VWD
VWF is synthesized in endothelial cells and becomes a multimer (formations of multiple molecules or monomers) that is held together by disulfide bridges.1 These multimers circulate in the plasma in an inactive form, only interacting when vascular damage occurs. At the time of vascular damage, VWF will bind to the exposed subendothelial collagen at the damaged vascular site, which allows the binding of platelets at the site and the binding of platelet collagen receptors to the collagen. This causes platelet adherence and aggregation of activated platelets, which inhibits further bleeding. In healthy individuals, VWF circulates throughout the body.1 Some individuals have mildly reduced circulating VWF not caused by a gene defect. If these people also have a bleeding tendency, then they are classified as having low VWF but not VWD. The disease itself is classified as either inherited or acquired on the basis of the underlying cause of disease.
Inherited VWD
Inherited VWD is classified into 3 main types on the basis of phenotypic, laboratory, and genetic factors.1,3 Type 1 is an autosomal dominant disease (i.e., inherited from 1 parent) characterized by a quantitative partial deficiency of VWF. Most patients have missense mutations, also known as point mutations, that exert dominance in the multimer. The mutations may be caused by impaired intracellular routing, storage, and excretion of VWF or accelerated clearance of VWF. Type 1 disease may also be caused by null alleles, mutant genes lacking normal gene function, which ultimately causes reduced synthesis of normal VWF.
Type 2 is also typically an autosomal dominant disease, and it is characterized by a dysfunctional VWF that causes decreased function of VWF.1,3 In Type 2 disease, missense mutations are the primary cause of disease. These mutations are typically associated with specific functional domains of VWF, and the specific mutations allow Type 2 disease to be further subdivided into 4 subtypes: 2A, 2B, 2M, and 2N. In Type 2A, these mutations cause defects in the multimerization, dimerization, or enhanced proteolysis of VWF. Type 2B is associated with enhanced spontaneous binding of glycoprotein Ib, which results in decreased platelet adherence. Type 2M, on the other hand, is characterized by reduced binding of VWF to glycoprotein Ib, resulting in decreased platelet adhesion and collagen binding. Type 2N is an autosomal recessive trait (i.e., inherited from both parents): an individual may either have 2 mutated 2N genes or 1 type 1 defect and 1 type 2N defect. Either of these combinations can cause decreased FVIII binding.
Type 3 disease is an autosomal recessive disorder, most often associated with null alleles, that results in the complete absence of VWF.1,3 Mutations may also occur in the promoter region of the gene. Some patients originally diagnosed with Type 3 disease are later discovered to have rapid clearance, rather than a complete absence, of VWF. Thus, they have only low levels of VWF and are reclassified as having Type 1 disease.4
Acquired VWD
Acquired VWD is characterized by a bleeding tendency that manifests in the presence of other conditions or diseases, including autoimmune disorders, cardiovascular disease, drugs, lymphoproliferative disorders, myeloproliferative disorders, solid tumors, and other conditions (Table 1).5,6 Typically, acquired VWD is also associated with mucosal manifestations of bleeding and post–trauma or surgery bleeding due to a reduction in VWF.
| Table 1. Conditions Associated with Acquired von Willebrand Disease5,6* |
| Underlying condition |
Examples |
| Autoimmune disorders |
- Graft-versus-host disease
- Hypothyroidism
- Other connective tissue disorders
- Systemic lupus erythematosus
|
| Cardiovascular diseases (acquired and inherited) |
- Aortic stenosis
- Atrial septal defects
- Angiodysplasia
- Endocarditis
- Mitral valve prolapse
- Systemic atherosclerosis
- Ventricular septal defects
|
| Drugs and therapeutic agents |
- Antibiotics (ciprofloxacin, griseofulvin, tetracycline)
- Hydroxyethyl starch
- Recombinant factor VIII
- Valproic acid
|
| Lymphoproliferative disorders |
- Acute lymphoblastic leukemia
- Chronic lymphocytic leukemia
- Hairy cell leukemia
- Non-Hodgkin lymphoma
- Monoclonal gammopathy of undetermined origin
- Multiple myeloma
- Waldenstrom macroglobulinemia
|
| Myeloproliferative disorders |
- Chronic myeloid leukemia
- Essential thrombocythemia
- Polycythemia vera
- Spontaneous myelofibrosis
|
| Non-hematologic malignancies |
- Primitive neuroectodermal tumors
- Renal cell carcinoma
- Solid tumors
- Wilms' tumor
|
| Other |
- Cirrhosis
- Diabetes mellitus
- Ehlers-Danlos syndrome
- Hemoglobinopathies
- Infections (e.g., pancreatic, viral)
- Myelodysplastic syndrome
- Lactoferrin deficiency
- Sarcoidosis
- Telangiectasia
- Turner syndrome
- Ulcerative colitis
- Uremia
|
| *Adapted with permission (creative commons license CC-BY-NC-ND 4.0). |
The etiology of acquired VWD is complex and may not be fully understood for all accompanying conditions. However, a number of mechanisms have been defined. For example, autoimmune diseases and lymphoproliferative disorders (e.g., lymphoma, myeloma) are typically associated with an immune–mediated reduction in VWF. The development of autoantibodies to VWF can lead to functional interference and, thus, bleeding. Patients with lymphoproliferative disorders, myeloproliferative disorders, and renal cancer can experience deficiencies because of the absorption of VWF on the abnormal or cancerous cells, resulting in enhanced clearance.5,6 This same mechanism is responsible in aortic valve stenosis, whereby high shear stress causes activation of thrombocytes and absorption of VWF. Also, mechanical or proteolytic consumption of or damage to VWF can occur, such as with myeloproliferative disorders, cardiovascular disease, uremia, and drugs such as ciprofloxacin.5
EPIDEMIOLOGY OF VWD
VWD is the most common inherited blood disorder, but, overall, it is quite rare. Based on epidemiologic studies, it is estimated to have a prevalence of up to 1.3%.1 However, because not all people with low VWF levels have clinically significant bleeding symptoms and the diagnostic cut–off level for “normal” VWF varies, the overall clinically relevant prevalence ranges from 0.005% to 0.1% in the literature.1,3,7 VWD is diagnosed most often in females because of menstrual bleeding and pregnancy–related bleeding risks.
The most common type of inherited VWD is Type 1; it accounts for 70% to 80% of all cases of inherited disease.1 Type 2 VWD occurs in approximately 20% of patients with the inherited form of the disease. Data on the frequency of the various subtypes are minimal. Data from France and Germany suggest that Types 2A (30% and 74%, respectively), 2B (28% and 10%, respectively), and 2N (34% and 3.5%, respectively), are the most common, while 2M is least common (8% and 13%, respectively).8 Type 3 disease, the most severe variant, occurs in less than 5% of patients with inherited VWD.
The prevalence of acquired VWD is unknown because of the lack of epidemiologic studies.6 Among all patients with VWD, it has been reported that 5% to 20% have acquired disease.6 Based on this data, the estimated prevalence in the entire population is 0.04% to 0.2%.6 Most commonly, acquired VWD is associated with lymphoproliferative disorders (48%), cardiovascular conditions (21%), and myeloproliferative disorders (15%).6 Less commonly, it is associated with cancers (5%), immunologic disorders (2%), and other miscellaneous conditions (9%).6
DIAGNOSIS OF VWD
VWD is diagnosed on the basis of a history of bleeding, as well as laboratory tests showing abnormalities in VWF and/or FVIII.1,7
Bleeding assessment
Histories of bleeding for both the patient and family members are the first assessment in the diagnostic work–up for VWD.1 Bleeding history has been shown to be highly specific (> 95%), but not highly sensitive (50%–60%), for the presence of disease.7 The personal history should include information about excessive bleeding, including spontaneity, severity, causes, sites, and duration.9 It should also include information about accompanying injuries and how easily the bleeding stopped. Family history is also important, as well as information about past personal medical history (i.e., other diseases that could contribute to bleeding) and medications (e.g., anticoagulants, nonsteroidal anti–inflammatory drugs [NSAIDs]). For people with VWD, the most common sites of bleeding are the nose, skin bruises, and menorrhagia or postpartum bleeding in women. Most bleeding is mild to moderate and does not require a physician visit. However, when a physical examination is completed as part of a diagnostic work–up or in cases of acute bleeding, the size, location, and distribution of ecchymoses, hematomas, petechiae, signs of anemia, or other causes of bleeding should be identified.
A bleeding score can be calculated to aid in the diagnosis of VWD: the score is the sum of the severities of all reported bleeding symptoms graded according to a scale.10 Recently, the International Society of Thrombosis and Haemostasis developed and endorsed a bleeding assessment tool for this calculation (https://bh.rockefeller.edu/ISTH–BATR/).11 When at least 3 different hemorrhagic symptoms are reported or the bleeding score is greater than 3 in males or greater than 5 in females, VWD is likely.7 However, such tools are limited in their diagnostic abilities; they are unsuitable for young children who have had no dental interventions or surgery, and severe bleeding episodes elevate the score, even if bleeding does not occur after a procedure.1 Therefore, these tools cannot be used alone to diagnose VWD.
Laboratory assessment
No single laboratory test can diagnose VWD. Instead, diagnosis requires the results of several tests that assess the pleiotropic function of VWF.1,7 A 2–tiered approach is often used for the diagnosis (Figure 1).1 The first level of tests include measurements of VWF antigen (VWF:Ag), VWF–dependent platelet adhesion (by the VWF–ristocetin cofactor activity assay [VWF:RCo]), and FVIII coagulant activity (FVIII:C).1 VWF:Ag levels are normally between 50 and 200 international units (IU)/dL9; VWF:RCo also typically ranges from 50 to 200 IU/dL. FVIII:C is considered normal if the level is within 50% to 150% of normal range, which varies with age. When all of these laboratory tests are normal, then VWD can be ruled out. However, if results are on the low end of normal, repeat testing may be warranted. If any abnormality in the first–level tests is present, then a diagnosis of VWD may occur. However, if the first–level test results are inconclusive, then second–level tests are performed. When VWF is less than 5 IU/dL, a diagnosis is made for Type 3 disease.1 However, this may inadvertently include some patients with severe Type 1 disease, so further second–level tests are needed before a firm diagnosis can be made.
When VWF is greater than 5 IU/dL, it must be considered in relation to the VWF:RCo. When this factor is less than 30 IU/dL, then the ratio of VWF:RCo to VWF:Ag needs to be calculated to differentiate between Types 1, 2A, 2B, and 2M disease (Figure 1). If this ratio is greater than 0.6, Type 1 disease is diagnosed; if the ratio is lower than 0.6, Type 2A, 2B, or 2M disease is present. An additional test will differentiate these 3 subtypes. Typically, FVIII:C and VWF:Ag levels are similar. However, in Type 2N, the reduction of FVIII:C is greater than the reduction of VWF:Ag (i.e., the ratio of FVIII:C to VWF:Ag ≤ 0.6).
The second–level tests include VWF propeptide (VWFpp), multimers, ristocetin–induced platelet aggregation (RIPA), and VWF–to–factor VIIIB (VFW:FVIIIB) binding activity, which, in combination with the first–level tests, can help distinguish between the subtypes of VWD (Figure 1).1 Propeptide, a cleavage product formed during the synthesis of VWF, is useful for distinguishing between Type 3 and Type 1 disease. In both types, VWF:Ag levels are very low (< 5 IU/dL). In fact, VWF:Ag may be completely absent in Type 3 disease. VWFpp levels are normal or reduced in Type 1, but VWFpp is absent in Type 3 disease. Recall that during the production of VWF, high molecular weight multimers are produced that are eventually cleaved into smaller multimers. With the loss of high molecular weight multimers, a decrease in platelet adhesion occurs. This deficiency in high molecular weight multimers occurs only in patients with Types 2A and 2B disease. The use of RIPA is used to further define the phenotypic changes observed in Types 2A and 2B. In Type 2A, RIPA is normal or reduced; in Type 2B, it is enhanced. To confirm that Type 2N disease is not hemophilia, a second–level test of the VFW:FVIIIB ratio can be conducted: the ratio should also be reduced if the patient has Type 2N VWD.
| Figure 1. Laboratory Diagnosis Algorithm for von Willebrand Disease1 |
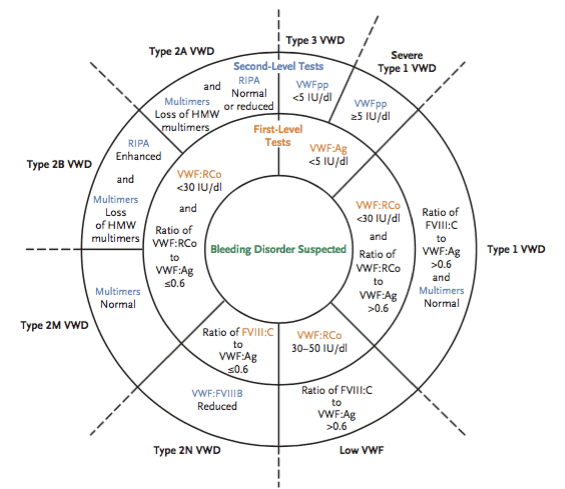 |
| Abbreviations: FVIII:C, factor VIII coagulant activity; HMW, high molecular weight; RIPA, ristocetin-induced platelet aggregation; VWD, von Willebrand disease; VWF:Ag, von Willebrand factor antigen; VWF:FVIIIB, von Willebrand factor¬–factor VIII binding activity; VFWpp, von Willebrand factor propeptide; VWF:RCo, von Willebrand factor¬–ristocetin cofactor activity. |
Although the diagnosis of VWD Types 2 and 3 is straightforward, Type 1 disease is a bit more controversial.1 Patients with Type 1 disease typically have a partial reduction in VWF, but the appropriate cut–off level for diagnosis is unknown.1,12 Current guidelines suggest that if a patient has a bleeding tendency and a VWF:RCo level between 30 and 50 IU/dL, then he either has possible Type 1 disease or simply low VWF levels. Another complicating factor is that VWF levels increase with increasing age. Patients may no longer meet diagnostic criteria for VWD even though they may still be at risk for bleeding as they age. With advances in technology and better categorization of the disease, some of these controversies may be resolved in the near future. Additionally, newer assays are being developed to speed the diagnostic process by combining several assays into a single test, and genotyping innovations are making this approach easier, as well.
TREATMENT OF VWD
The goal of VWD treatment is to normalize VWF and FVIII levels in case the patient bleeds or needs an interventional procedure with a risk of bleeding, such as surgery. Typically, the first treatment strategy is to use desmopressin to facilitate release of endogenous VWF stores, which, in turn, increases plasma concentrations of VWF.8 Second, plasma–derived, viral–inactivated, or recombinant replacement factor concentrates can be used. A third strategy, using antifibrinolytics, is aimed at promoting hemostasis and wound healing but does not alter VWF concentrations. The appropriateness of one strategy over another depends on the type and severity of VWD, the severity of hemostatic challenge, and the type of actual or potential bleeding.
Desmopressin
Desmopressin is a synthetic derivative of the antidiuretic hormone L–argine vasopressin and is an effective prohemostatic agent in patients with bleeding disorders.13,14 It has been used in the treatment of both VWD and hemophilia since the late 1970s.13 Desmopressin acts through type 2 vasopressin receptors to stimulate the secretion of VWF and tissue plasminogen activator from endothelial cells to plasma, enhancing the binding to platelets. FVIII is also released from storage sites following desmopressin administration and the VWF protects it from degradation, contributing to its sustained activity. Furthermore, desmopressin also increases glycoprotein Ib/IX and CD62 expression, which improves adhesion of platelets. Each of these actions contributes to shortened bleeding time and normalizing platelet aggregation.
Desmopressin is currently available in intravenous (IV), subcutaneous, intranasal, and oral formulations.13 However, the oral formulation is not effective in patients with bleeding disorders. In patients with VWD, the high concentration (1.5 mg/mL) of the intranasal solution (Stimate) should be used, not the lower–concentration (0.01%) formulation. The intranasal route is associated with much more variability in pharmacokinetics, including absorption, time to peak effect, and duration of effect. Generally, responses are observed within 1 to 4 hours and last up to 8 hours. Any nasal obstruction or irritation, though, can decrease absorption. The IV formulation increases FVIII:C 2– to 4–fold within 30 minutes in patients with bleeding disorders; levels return to baseline within 24 hours. Desmopressin also increases VWF levels, but not as dramatically, and response varies among patients. The subcutaneous formulation is similar in its effect to the IV form, but it takes slightly longer to achieve peak activity (i.e., 60–90 minutes compared to 30–60 minutes with IV).
Desmopressin is most effective in Type 1 VWD, in those with functionally normal VWF, and in children older than 2 years of age.13 Typically, 80% to 90% of patients will respond to desmopressin treatment. Children younger than 2 years old have decreased responsiveness but become more responsive as they age. Response is often defined as increases in FVIII and VWF levels of at least 2– to 3–fold within 2 hours of administration. Because many factors influence dose response, establishing the magnitude and duration of response is important.1 Before the first dose, pre–infusion laboratory testing and repeat testing at 1, 2, and/or 4 hours should be completed to assess FVIII concentrations, VWF:Ag, and VWF:RCo.17 Sometimes, this is referred to as the “test dose” because it tests the effect of desmopressin, although the dose used for the test is the same as the standard dose.
The standard IV or subcutaneous dose of desmopressin is 0.3 mcg/kg; it is typically prepared in 50 mL of normal saline (unless body weight is less than 10 kg) and administered over 30 minutes, with a maximum dose of 20 mcg to avoid side effects.13,15 The inhaled dose of the high–concentration (1.5 mg/mL) nasal formulation is 150 mcg (1 puff) for patients weighing 50 kg or less and 300 mcg (2 puffs) for those weighing more than 50 kg. Repeated dosing can lead to tachyphylaxis, although this risk is more pronounced in those with hemophilia. Second and subsequent doses are approximately 30% less effective than the first dose. Therefore, repeat dosing should occur at 24–hour intervals when administered for more than 2 to 4 days.
Side effects of desmopressin are typically mild to moderate in nature and are transient, usually lasting no more than 24 hours.13,14 The incidence of side effects is variable and dependent on the route of administration: the IV route is associated with the most side effects. Vasomotor–type effects of desmopressin can cause headache, facial flushing, mild hypotension, and tachycardia. These effects are most common with IV administration and can be attenuated with slowing the infusion. Mild hyponatremia (< 135 mmol/L) can occur, but severe hyponatremia and related effects (e.g., seizures) are rare, except in children younger than 2 years old, with repetitive dosing, or with aggressive fluid replacement during surgery. Risks of uterine or gastrointestinal contractions and hypertension are rare because desmopressin only minimally affects type 1 vasopressin receptors. In elderly people with cardiovascular or cerebrovascular risk factors, rare instances of thrombotic events have occurred following the administration of desmopressin. The product labeling recommends avoiding desmopressin in patients with uncontrolled hypertension or these risk factors. Both blood pressure and heart rate should be monitored during administration of the IV formulation.15
Factor concentrate
When desmopressin is ineffective, causes an inadequate response, or is contraindicated, factor replacement therapy is used to treat bleeding. In fact, for patients with Type 3 and most patients with Type 2 VWD, factor concentrate is the recommended treatment choice.1,16 It is also for Type 1 patients who have failed desmopressin or who have contraindications to desmopressin. Both plasma–derived and recombinant products are commercially available in the United States (U.S.). Although products are available that primarily contain FVIII and little or no VWF, these are generally not useful in patients with VWD. Plasma–derived concentrates include both FVIII and VWF in various ratios; the available recombinant formulation only includes VWF (Table 2).17 The plasma–derived factor concentrates vary in terms of relative proportions of the factors, and this may have implications for treatment, because the plasma–derived factor concentrates are not considered interchangeable (Table 2).8,17–21 Furthermore, production techniques can cause variable amounts of high molecular weight VWF, which are the most adhesive and functional forms of the factor and, in theory, a concentrate containing these forms could be more efficacious than concentrates containing less functional forms.8 Recombinant factor concentrate contains these high molecular weight VWF forms, but the plasma–derived factor concentrates are variably deficient in the high molecular weight forms (Table 2). The plasma–derived factor concentrates are derived from plasma collected from paid donors, which confers a small risk of viral transmission. Recombinant factor concentrate does not carry this risk. Neutralizing antibodies can form with any of the plasma–derived or recombinant factor concentrates; however, no reports of neutralizing antibodies with the recombinant concentrate have been reported. Despite the advantages of recombinant factor concentrate, the clinical use of this concentrate is relatively new and the best uses of it in special populations, such as those with major gastrointestinal bleeding, pregnant women, and children, are still being evaluated.
| Table 2. Characteristics of Currently Available Factor Concentrates16-21 |
| Characteristic |
Alphanate |
Humate-P |
Vonvendi |
Wilate |
| Factor type |
VWF/FVIII concentrate |
VWF/FVIII concentrate |
Recombinant VWF |
VWF/FVIII concentrate |
| VWF specific activity (U/mg protein) |
≥100 |
≥80 |
N/A |
70 ± 30 |
| VWF:RCo/VWF:Ag (ratio) |
0.43-0.9 |
0.8-0.91 |
>1 |
0.9-1.0 |
| VWF:RCo/FVIII:C (ratio) |
0.82-1.2 |
2.4-2.88 |
N/A |
1.0 |
| High molecular weight multimers |
Variably deficient |
Variably deficient |
Present |
Variably deficient |
| Carbohydrate structure/glycosylation of VWF |
Normal |
Normal |
Altered? |
Normal |
| Other components |
Albumin |
Fibrinogen, albumin |
None |
None |
| Median half-life of VWF:RCo/FVIII:C (hours) |
6.91 |
10.3-11.3 |
19.1-21.9 |
9.1-24.7 |
| FDA-approved indication(s) |
Surgical and/or invasive procedures in adult and pediatric patients with VWD in whom desmopressin is ineffective or contraindicated (except patients with Type 3 disease);
control and prevention of bleeding in patients with hemophilia A or acquired FVIII deficiency |
Treatment of spontaneous and trauma-induced bleeding episodes or prevention of excessive bleeding during and after surgery in adult and pediatric patients with VWD; treatment and prevention of bleeding in adults with hemophilia A |
Treatment and control of bleeding episodes in adults with VWD |
Control of bleeding and perioperative management of bleeding in children and adults with VWD |
| Contraindications |
Life-threatening hypersensitivity reactions to product or components (albumin, calcium, glycine, heparin, histidine, imidazole, arginine, polyethylene glycol and polysorbate 80, sodium tri-n-butyl phosphate) |
Life-threatening hypersensitivity reactions to product or components |
Life-threatening hypersensitivity reactions to recombinant VWF or components (mannitol, trehalose, sodium chloride, histidine, Tris, calcium chloride, polysorbate 80, hamster or mouse proteins) |
Life-threatening hypersensitivity reactions to product or components (glycine, sucrose, sodium chloride, sodium citrate, calcium chloride, polysorbate 80) |
| Warnings |
- Anaphylaxis and severe hypersensitivity reactions may occur
- Neutralizing antibodies may develop (occasional reports in Type 3 patients)
- Thromboembolic reactions may occur, especially in those with increased risk factors for thrombosis
- Intravascular hemolysis may occur with massive doses
- Vasomotor reactions may occur with rapid administration
- Virus transmission is possible
|
- Thromboembolic events may occur
- Intravascular hemolysis may occur; monitor for decreasing hematocrit values in patients with A, B, and AB blood groups receiving large or frequent doses
- Monitor VWF:RCo and FVIII levels, especially in those undergoing surgery
- Virus transmission is possible
|
- Thromboembolic reactions may occur, especially in those with increased risk factors for thrombosis; monitor FVIII with frequent dosing
- Hypersensitivity reactions may occur
- Neutralizing antibodies may develop (if expected VWF:RCo not achieved and bleed not controlled, assess for antibodies)
|
- Anaphylaxis and severe hypersensitivity reactions may occur
- Neutralizing antibodies may develop, especially in Type 3 patients
- Thromboembolic reactions may occur, especially in those with increased risk factors for thrombosis; monitor plasma levels of FVIII
- Virus transmission is possible
|
| Abbreviations: FDA, United States Food and Drug Administration; FVIII, factor VIII; FVIII:C factor VIII coagulant activity;
N/A, not applicable; VWD, von Willebrand disease; VWF, von Willebrand factor; VWF:Ag, von Willebrand factor antigen;
VWF:RCo, von Willebrand factor–ristocetin cofactor activity. |
Studies evaluating acute spontaneous bleeding and bleeding after surgical events show that the use of plasma–derived replacement therapy produces very good to excellent results, with 86% to 100% of patients achieving bleeding control.16 In surgery and major bleeding events, response rates of 93% to 100% have been reported.8 Recombinant VWF produces similar results. In a phase III study of VWD patients, bleeding control was rated as “excellent” in 97% of the 192 bleeds in 22 patients, and a single infusion resulted in a bleeding control rate of 81.8%.22 In this phase III study, the FVIII:C increased rapidly (i.e., within 6 hours after the infusion) and was sustained for 72 hours post–infusion.
The dose and duration of treatment with factor concentrates depend on the type of bleeding (mild–moderate or severe) and the type of intervention (Table 3).1 The dose of factor concentrate depends on the type of concentrate and the specific brand, especially if it contains both FVIII and VWF. Essentially, the dose is based on the anticipated recovery of factors and target levels of both VWF:RCo and FVIII:C. The expectation is that each unit of FVIII infused per kilogram of body weight should increase FVIII:C by 2 IU/dL and that every unit of VWF:RCo infused per kilogram of body weight should increase VWF:RCo activity by 1.5 IU/dL.1 It is also important to note that, if using recombinant VWF concentrate, a single–dose of FVIII is necessary to prompt immediate hemostasis.1
Monitoring of the VWF:RCo and FVIII:C levels is critical to ensuring that the adequate amount and duration of replacement have occurred.1,8 Typically, the VWF:RCo trough and FVIII:C peak should be monitored daily, but the length of monitoring is dependent on the type of bleed, procedure, or surgery (Table 3). It is important to note that exceeding a VWF:RCo level of 200 IU/dL and an FVIII:C level of 250 to 300 IU/dL can increase thrombotic risk. In some cases, clinicians may opt for alternating factor replacement therapy with desmopressin during the latter portion of treatment to minimize this risk.17
| Table 3. Dosing Regimens for Factor Concentrates1 |
| Bleeding indication |
Doseb
(IU/kg) |
Monitoring |
Treatment duration
(days) |
Comments |
| Bleeding |
|
|
|
- Treat in centers that have extensive experience and access to local hemostasis lab
|
| Mild to moderate |
20-40 |
Peak VWF:RCo > 50-80 IU/dL on day 1
Trough FVIII > 30 IU/dL after day 1 |
1-3 |
| Severe |
50 |
Peak VWF:RCo > 100 IU/dL on day 1
Trough FVIII > 50 IU/dL after day 1 |
7-10 |
Procedurea
- Cardiac catheterization
- Cataract surgery
- Dental extraction (simple)
- Endoscopy (no biopsy)
- Laceration repair
- Liver biopsy
|
25 |
Peak VWF:RCo > 50 IU/dL on day 1 |
1 |
- Treat in centers that have extensive experience and access to local hemostasis lab
|
Minor surgerya
- Biopsy (breast, cervical)
- Central line placement
- Dental extractions (complicated)
- Gingival surgery
- Laparoscopic procedures
|
30-60 |
Peak VWF:RCo > 50-80 IU/dL on day 1
Trough FVIII > 30 IU/dL after day 1 |
1-5 |
- Treat in centers that have extensive experience and access to local hemostasis lab
- Monitor trough levels regularly to maintain FVIII > 50 IU/dL x 1-5 d
- If measurement of VWF:RCo not immediately available, dose based on FVIII:C alone
|
Major surgerya
- Cardiothoracic
- Cesarean delivery
- Craniotomy
- Hysterectomy
- Open cholecystectomy
- Prostatectomy
|
50-60 |
Peak VWF:RCo > 100 IU/dL on day 1
Trough FVIII > 50 IU/dL after day 1 |
7-10 |
- Treat in centers that have extensive experience and access to local hemostasis lab
- Both values should be > 100 IU/dL to ensure normal hemostasis; monitor trough levels regularly to maintain > 50 IU/d x 7-10 d
- If measurement of VWF:RCo not immediately available, dose based on FVIII:C alone
|
| Delivery |
40-50 |
Peak VWF:RCo > 100 IU/dL on day 1
Trough FVIII > 50 IU/dL after day 1 |
3-4 |
- Treat in centers that have extensive experience and access to local hemostasis lab
|
Abbreviations: FVIII, factor VIII; FVIII:C, factor VIII coagulant activity; VWF, von Willebrand factor; VWF:RCo, von Willebrand factor–ristocetin cofactor activity.
aExample procedures or surgeries.
bDepends on type and brand of concentrate used. If recombinant VWF is used, then a single dose of FVIII concentrate should be administered to achieve an immediate FVIII target level. |
Side effects from replacement factor concentrate therapy are rare but may include allergic and anaphylactic symptoms, particularly with plasma–derived products.8 If these reactions occur, the infusion should be stopped immediately and appropriate allergic/anaphylactic treatment should be administered. If factor replacement concentrate is needed again, consideration of an alternative agent (e.g., a recombinant product) is suggested. High levels of FVIII increase the risk of thrombosis and a warning of this risk has been placed on all of the plasma–derived and recombinant factor product labeling.18–21 Of note, in the pivotal phase III trial of recombinant VWF, no anaphylaxis or thrombotic events occurred.22 However, no patients with a history of thromboembolic disease were enrolled in this study, which is not fully representative of the population of patients that will use these products. Therefore, it is prudent, regardless of product used, to use caution and carefully monitor patients during replacement therapy to minimize the risk of thrombosis, if patients have known risk factors for thromboembolism, including older age, previous thrombosis, obesity, surgery, immobility, hormone replacement therapy, and use of fibrinolytic therapy.16
Antifibrinolytic treatment
Antifibrinolytic treatments, including aminocaproic acid and tranexamic acid, can be quite useful, particularly for patients with mucocutaneous bleeding.1 These agents can also be used to reduce bleeding or prevent re–bleeding in patients undergoing surgery or dental procedures. Both drugs exert their actions by inhibiting the conversion of plasminogen to plasmin, ultimately inhibiting fibrinolysis and stabilizing clot formation. These drugs are currently available in both IV and oral formulations and are approved by the U.S. Food and Drug Administration for multiple bleeding–related indications (e.g., menstrual bleeding, hemophilia).23–25
Data from randomized clinical trials are lacking to support the use of antifibrinolytics in bleeding disorders, but anecdotal evidence has shown benefit of antifibrinolytics in some patients, and data from 2 trials performed in patients with hemophilia have supported their use. The 2 trials of hemophilia patients, which were published in the early 1970s, reported beneficial effects of systemic tranexamic acid and aminocaproic acid, including reductions in number of bleeds, amount of blood lost, and need for therapeutic factor replacement concentrate.26 In these studies, side effects were rare and led to discontinuation of therapy in only 1 patient.
Dosing of IV aminocaproic acid is initiated with 4 to 5 g as a loading dose 1 hour before an invasive procedure, and then 1 g per hour every 4 to 6 hours until bleeding is controlled or for a total of 5 to 7 days.8 The daily oral dose is similar: 4 to 5 g as a loading dose and then 4 to 6 g every 4 to 6 hours. The total daily dose should be limited to 24 g to minimize side effects, which are mainly gastrointestinal in nature. For children, weight–based dosing of 50 to 60 mg/kg is used. Lower doses of 25 mg/kg are still effective and can be used if gastrointestinal side effects occur.
The dose of tranexamic acid is 10 to 15 mg/kg every 8 hours administered IV or orally. Before an oral dosage form was available in the U.S., the IV formulation was used as an oral rinse (e.g., swish and swallow approach) for mucocutaneous bleeding. However, this is not an FDA–approved indication for this formulation. Compounded mouthwashes have shown effectiveness in small studies,27 but none are commercially available.
Because antifibrinolytics are excreted renally, dose adjustments are needed in patients with renal insufficiency.8 Side effects of these therapies include nausea and vomiting and, less frequently, thrombotic events.8 Contraindications to antifibrinolytic treatment include disseminated intervascular coagulation and bleeding from renal parenchyma or the upper urinary tract because these agents can cause renal clots or obstruction leading to renal failure.
Prophylactic treatment
Unlike with hemophilia, prophylactic infusion of factor concentrate or administration of desmopressin to prevent bleeding is not commonly employed in patients with VWD, although some benefit has been reported.1,28,29 The results of 1 retrospective study demonstrated efficacy in reducing annualized bleeding rates, but cost effectiveness of this approach has not been proven.30 A recent prospective study of 11 patients with severe VWD showed promising results.30 In the study, patients were administered VWF:RCo at a dose of 50 IU/kg 2 or 3 times weekly. Reductions in number of bleeding episodes, gastrointestinal and joint bleeding, and severe epistaxis were noted, but slow accrual of participants caused the study to close. Additionally, in women with severe VWD, prophylaxis may be used for a short time during the postpartum period.31
Women's health and treatment of VWD
Pregnancy–related and menstrual bleeding pose considerable challenges for women with VWD.
Menorrhagia
In some cases, menorrhagia can be the first sign of VWD, and, according to some reports, up to 20% of women with menorrhagia have VWD.9,32 Whether it is a first sign or not, though, menorrhagia needs to be appropriately managed in women with VWD. After ruling out gynecologic or hormonal causes of menorrhagia, treatment depends on a woman's desire to get pregnant. Women not desiring a pregnancy at the time of diagnosis can be treated with an oral contraceptive containing both estrogen and progestin. Alternatively, a levonorgesterol–releasing intrauterine device, such as Mirena, can be used. In women wishing to consider pregnancy, management with desmopressin, antifibrinolytics, or factor concentrate replacement can be used. Referral to a hemophilia center is recommended, regardless of the treatment selected, because these patients are at high risk for bleeding.
Desmopressin can be self–administered subcutaneously or intranasally by a woman at the onset of menses. When administered subcutaneously every 12 hours beginning on day 1 of menses and repeated 24 hours later, an 86% response rate (i.e., control of bleeding described by patient using a questionnaire and direct interview by healthcare professional) has been reported.33 The intranasal administration of the high–concentration formula (1.5 mg/mL) at doses of 150 mcg/spray/day in women weighing less than 50 kg and 300 mcg/spray/day in women weighing more than 50 kg (until bleeding is controlled) produced similar results (88% response rate).34 Women used desmopressin an average of 1.7 days per cycle (range, 1–8) and menses was slowed and lightened in most women. Tranexamic acid 15 to 25 mg/kg 3 times daily during the entire menstrual period can also be used.32 Factor concentrate can be used if desmopressin or tranexamic acid is not effective or is contraindicated. Iron therapy may be needed if bleeding continues to be severe or chronic to prevent iron–deficiency anemia. Hysterectomy or endometrial ablation can be used if these approaches are unsuccessful.1
Pregnancy
Although an increased risk of miscarriage or fetal harm has not been shown in women with VWD, they are at much higher risk of postpartum bleeding (30% vs 5% in general population).32 In healthy women, 2– to 3–fold increases in both VWF and FVIII levels occur during pregnancy and return to baseline following delivery.1 This also is observed in women with Type 1 VWD. Women with Type 2 VWD may also experience a rise in FVIII antigen, although VWF remains reduced because of the defects observed in proteins. Furthermore, those with Type 2N disease show a complete normalization of FVIII:C and often do not require treatment. Nonetheless, all women with VWD who are pregnant should be closely monitored and VWF:RCo and FVIII:C should be measured during the third trimester, within 10 days of the expected delivery date, and for 2 weeks postpartum.
When both VWF:RCo and FVIII:C levels are 50 IU/dL or greater before delivery, spinal and epidural anesthesia is safe and the risks of bleeding are minimal.32 If levels are less than 50 IU/dL, then factor concentrate replacement is administered to prevent bleeding, with the goal of therapy being to increase levels to greater than 100 IU/dL by the time of delivery.1 In patients with Type 1 VWD, if FVIII:C levels are less than 50 IU/dL, desmopressin administration is necessary immediately after umbilical cord clamping and for up to 2 days following delivery.33 Levels usually return to baseline within 7 to 21 days following delivery.17 Desmopressin during pregnancy for other types of bleeding episodes is generally not recommended because of limited safety data.32 Tranexamic acid may be safely administered in the postpartum period, if needed. Limited data exist for desmopressin or tranaxemic acid in breastfeeding women. One study conducted in postpartum women found small amounts of desmopressin in breast milk.15 Tranexamic acid is present in breastmilk at 1 one–hundredth of the corresponding serum concentration.25 A risk/benefit discussion should occur with women who are using these medications and desire to breastfeed.
Treating acquired VWD
In patients with acquired VWD, treatment is first directed at controlling active bleeding, preventing future bleeding, and then achieving remission of the underlying disease.5,17 If the patient needs to undergo an invasive procedure, it is best to postpone the procedure, if possible, until the underlying disease is treated. Although similar treatment strategies to those used for inherited forms of VWD can be applied to patients with acquired disease, some may not work as well due to the mechanism of disease or the presence of alloantibodies. For example, the half–lives of VWF concentrate administered as replacement therapy and desmopressin may be shortened if the mechanism for acquired disease is an increased clearance of VWF.5,17
A registry of patients with acquired VWD has collected data on patients, which may help assess the usefulness of treatment strategies in acquired VWD. Overall, 32% of patients with acquired VWD respond to treatment with desmopressin. Those with autoimmune disorders, lymphoproliferative conditions, and non–hematologic cancers have the best prognosis.5 Patients receiving factor concentrate replacement should be monitored closely and the dose and frequency of administration should be based on clinical response.
The treatment for some patients, such as those with monoclonal gammopathy of uncertain significance, lymphoproliferative disease, autoimmune disease, and cancers, is immunoglobulin 1 g/kg daily for 2 days.5,17 VWF activity is observed 24 to 48 hours after administration of immunoglobulin. Thus, patients may need concomitant administration of desmopressin or factor concentrate replacement to stop active bleeding. Other options include plasmapheresis to eliminate autoantibodies and paraproteins and corticosteroids or immunosuppressants to block or eliminate autoantibodies.
If, despite these measures, bleeding remains uncontrolled, then administration of recombinant FVIII (90 mcg/kg) has been shown to be effective in patients with alloantibodies.8,17 Once bleeding is controlled, if underlying disease can also be controlled, the risk of subsequent bleeds is minimized.
PHARMACIST'S ROLE IN VWD
Pharmacists can play a pivotal role in managing patients with VWD in all health care settings. Pharmacists should review patients' medication lists throughout their courses of VWD to ensure that patients are not using aspirin or NSAIDs for pain. They should also ensure that other anticoagulants are not prescribed, unless a hematologist is involved with that decision. Pharmacists should educate patients about the appropriate use of preventative or therapeutic agents (e.g., desmopressin, factor replacement, antifibrinolytics); the need for the patient to wear or carry identifying information about their disease, as well as provider contact information, in the event of an emergency; and when and where to seek emergency care if a bleeding event happens. Also, pharmacists should work with patients to ensure that any drug therapy is affordable and taken appropriately to facilitate the best possible outcomes.
The hospital pharmacist can ensure that there are adequate supplies of factor concentrate, desmopressin, and antifibrinolytics in stock, particularly when VWD patients are undergoing planned procedures. Often, this requires pharmacists to be actively involved in justifying the high cost of these drugs for budget purposes and to have procedures in place to obtain factor concentrate quickly and minimize waste. Pharmacists should be involved in calculating or double–checking the calculations of the doses of factor concentrates, ensuring that the dosing is based on the VWF:RCo units and that both VWF:RCo and FVIII levels are monitored following administration. Pharmacists can have a critical role in following the laboratory results and monitoring for response and the need for subsequent doses.
Pharmacists in the outpatient or specialty settings are also responsible for ensuring appropriate care for VWD patients. These pharmacists have similar responsibilities to the hospital pharmacist for patients who receive treatment in the home care setting. They are also involved in educating not only the patient and caregivers about appropriate storage, reconstitution, and administration of therapies at home but also those in the community, including school personnel, local dentists, and home care nurses. Pharmacists in the community or specialty pharmacy settings can take special care to ensure that patients receive the high–concentration desmopressin nasal spray. Desmopressin and factor concentrates are often in the highest tier for insurance copays, which often necessitates financial assistance for patients, and pharmacists can assist with finding support for the patient. With such an interprofessional approach to managing VWD patients, optimal outcomes can be achieved.
REFERENCES
- Leebeek FW, Eikenboom JC. Von Willebrand's disease. N Engl J Med. 2016;375(21):2067–2080.
- James AH, Eikenboom J, Federici AB. State of the art: von Willebrand disease. Haemophilia. 2016;22 Suppl 5:54–59.
- Sanders YV, Groeneveld D, Meijer K, et al; WiN study group. von Willebrand factor propeptide and the phenotypic classification of von Willebrand disease. Blood. 2015;125(19):3006–3013.
- Federici AB. Current and emerging approaches for assessing von Willebrand disease in 2016. Int J Lab Hematol. 2016;38 Suppl 1:41–49.
- Mital A. Acquired von Willebrand syndrome. Adv Clin Exp Med. 2016;25(6):1337–1344.
- Sucker C, Michiels JJ, Zotz RB. Causes, etiology and diagnosis of acquired von Willebrand disease: a prospective diagnostic workup to establish the most effective therapeutic strategies. Acta Haematol. 2009;121(2–3):177–182.
- Castaman G, Linari S. Diagnosis and treatment of von Willebrand disease and rare bleeding disorders. J Clin Med. 2017;6(4).pii:E45.
- The diagnosis, evaluation, and management of von Willebrand disease. NIH Publication No. 08–5832. U.S. Department of Health and Human Services; National Institutes of Health; National Heart, Lung, and Blood Institute. https://www.nhlbi.nih.gov/files/docs/guidelines/vwd.pdf. Published December 2007. Accessed September 6, 2017.
- Yawn BP, Nichols WL, Rick ME. Diagnosis and management of von Willebrand disease: guidelines for primary care. Am Fam Physician. 2009;80(11):1261–1268.
- Tosetto, A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM–1 VWD). J Thromb Haemost. 2006;4(4):766–773.
- Rodeghiero F, Tosetto A, Abshire T, et al; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065.
- Bowman ML, James PD. Controversies in the diagnosis of type 1 von Willebrand disease. Int J Lab Hematol. 2017;39 Suppl 1:61–68.
- Leissinger C, Carcao M, Gill JC, et al. Desmopressin (DDAVP) in the management of patients with congenital bleeding disorders. Haemophilia. 2014;20(2):158–167.
- Stoof SC, Cnossen MH, de Maat MP, et al. Side effects of desmopressin in patients with bleeding disorders. Haemophilia. 2016;22(1):39–45.
- DDAVP injection [prescribing information]. Bridgewater, NJ: Sanofi–Aventis US, LLC;2007.
- Franchini M, Mannucci PM. Von Willebrand factor (Vonvendi®): the first recombinant product licensed for the treatment of von Willebrand disease. Expert Rev Hematol. 2016;9(9):825–830.
- Curnow J, Pasalic L, Favaloro EJ. Treatment of von Willebrand disease. Semin Thromb Hemost. 2016;42(2):133–146.
- Alphanate [prescribing information]. Los Angeles, CA: Grifols Biologicals, Inc.;2015.
- Humate–P [prescribing information]. Kankakee, IL: CSL Behring GmbH;2016.
- Vonvendi [prescribing information]. Westlake Village, CA: Shire;2015.
- Wilate [prescribing information]. Hoboken, NJ: Octapharma USA, Inc.;2015.
- Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombination von Willebrand factor in severe von Willebrand disease. Blood. 2015;126(17):2038–2046.
- Amicar [prescribing information]. Lake Forest, IL: Clover Pharmaceuticals Corp.;2017.
- Lysteda [prescribing information]. Parsippany, NJ: Ferring Pharmaceuticals, Inc.;2016.
- Cycklokapron [prescribing information]. New York, NY: Pfizer Inc.;2016.
- Van Galen KP, Engelen ET, Mauser–Bunschoten EP, et al. Antifibrinolytic therapy for preventing oral bleeding in patients with haemophilia or von Willebrand disease undergoing minor oral surgery or dental extractions. Cochrane Database Syst Rev. 2015;(12):CD011385.
- Nuvvula S, Gaddam KR, Kamatham R. Efficacy of tranexamic acid mouthwash as an alternative for factor replacement in gingival bleeding during dental scaling in cases of hemophilia: a randomized clinical trial. Contemp Clin Dent. 2014;5(1):49–53.
- Berntorp E, Petrini P. Long–term prophylaxis in von Willebrand disease. Blood Coagul Fibrinolysis. 2005;16 Suppl 1:S23–S26.
- Abshire T. The role of prophylaxis in the management of von Willebrand disease: today and tomorrow. Thromb Res. 2009;124 Suppl 1:S15–19.
- Abshire TC, Federici AB, Alvárez MT, et al; VWD PN. Prophylaxis in severe forms of von Willebrand's disease: results from the von Willebrand Disease Prophylaxis Network (VWD PN). Haemophilia. 2013;19(1):76–81.
- Abshire T, Cox–Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost. 2015; 13(9):1585–1589.
- Rodeghiero F. Von Willebrand disease: pathogenesis and management. Thromb Res. 2013;131 Suppl 1:S47–S50.
- Rodeghiero F, Castaman G, Mannucci PM. Prospective multicenter study on subcutaneous concentrated desmopressin for home treatment of patients with von Willebrand disease and mild or moderate hemophilia A. Thromb Haemost. 1996;76(5):692-696.
- Leissinger C, Becton D, Cornell C Jr, Cox Gill J. High–dose DDAVP intranasal spray (Stimate) for the prevention and treatment of bleeding in patients with mild hemophilia A, mild or moderate type 1 von Willebrand disease and symptomatic carriers of hemophilia A. Haemophilia. 2001;7(3):258–266.
Back to Top